

Food and Drug Regulations (C.R.C., c. 870)
Full Document:
- HTMLFull Document: Food and Drug Regulations (Accessibility Buttons available) |
- XMLFull Document: Food and Drug Regulations [4742 KB] |
- PDFFull Document: Food and Drug Regulations [13030 KB]
Regulations are current to 2024-11-26 and last amended on 2024-06-17. Previous Versions
Food and Drug Regulations
C.R.C., c. 870
Regulations Respecting Food and Drugs
PART AAdministration
General
A.01.001 These Regulations may be cited as the Food and Drug Regulations.
A.01.002 These Regulations, where applicable, prescribe the standards of composition, strength, potency, purity, quality or other property of the article of food or drug to which they refer.
A.01.003 [Repealed, SOR/94-289, s. 1]
Interpretation
A.01.010 In these Regulations,
- acceptable method
acceptable method means a method of analysis or examination designated by the Minister as acceptable for use in the administration of the Act and these Regulations; (méthode acceptable)
- Act
Act means the Food and Drugs Act, except in Parts G and J; (Loi)
- common-law partner
common-law partner has the same meaning as in section 2 of the Criminal Code; (conjoint de fait)
- cubic centimetre
cubic centimetre and its abbreviation cc. shall be deemed to be interchangeable with the term millilitre and its abbreviation ml.; (centimètre cube)
- Director
Director[Repealed, SOR/2018-69, s. 1]
- inner label
inner label means the label on or affixed to an immediate container of a food or drug; (étiquette intérieure)
- Lot number
Lot number means any combination of letters, figures, or both, by which any food or drug can be traced in manufacture and identified in distribution; (numéro de lot)
- manufacturer
manufacturer[Repealed, SOR/97-12, s. 1]
- manufacturer
manufacturer or distributor means a person, including an association or partnership, who under their own name, or under a trade-, design or word mark, trade name or other name, word or mark controlled by them, sells a food or drug; (fabricant or distributeur)
- official method
official method means a method of analysis or examination designated as such by Minister for use in the administration of the Act and these Regulations; (méthode officielle)
- outer label
outer label means the label on or affixed to the outside of a package of a food or drug; (étiquette extérieure)
- prescription drug
prescription drug means a drug that is set out in the Prescription Drug List, as amended from time to time, or a drug that is part of a class of drugs that is set out in it; (drogue sur ordonnance)
- Prescription Drug List
Prescription Drug List means the list established by the Minister under section 29.1 of the Act; (Liste des drogues sur ordonnance)
- principal display panel
principal display panel has the same meaning as in the Consumer Packaging and Labelling Regulations; (espace principal)
- security package
security package means a package having a security feature that provides reasonable assurance to consumers that the package has not been opened prior to purchase. (emballage de sécurité)
- SOR/84-300, s. 1(F)
- SOR/85-141, s. 1
- SOR/89-455, s. 1
- SOR/97-12, s. 1
- SOR/2000-353, s. 1
- SOR/2001-272, s. 5
- SOR/2003-135, s. 1
- SOR/2013-122, s. 1
- SOR/2018-69, ss. 1, 27
- SOR/2022-197, s. 1
A.01.011 The Minister shall, upon request, furnish copies of official methods.
- SOR/2018-69, s. 27
A.01.012 The Minister shall, upon request, indicate that a method is acceptable or otherwise upon its submission to him for a ruling.
- SOR/2018-69, s. 27
A.01.013 Where a food, drug, vitamin or cosmetic has more than one name, whether proper or common, a reference in these Regulations to the food, drug, vitamin or cosmetic by any of its names is deemed to be a reference to the food, drug or vitamin by all of its names.
A.01.014 When a lot number is required by these Regulations to appear on any article, container, package or label it shall be preceded by one of the following designations:
(a) “Lot number”;
(b) “Lot No.”;
(c) “Lot”; or
(d) “(L)”.
A.01.015 (1) Subject to subsection (2), any statement, information or declaration that is required by these Regulations to appear on the label of any drug shall be in either the French or the English language in addition to any other language.
(2) The adequate directions for use required to be shown on the inner and outer labels of a drug pursuant to subparagraph C.01.004(1)(c)(iii) shall be in both the French and English languages if the drug is available for sale without prescription in an open self-selection area.
- SOR/85-140, s. 1
A.01.016 All information that is required by these Regulations to appear on a label of a food or a drug, other than a drug for human use in dosage form, shall be
(a) clearly and prominently displayed on the label; and
(b) readily discernible to the purchaser or consumer under the customary conditions of purchase and use.
- SOR/2014-158, s. 1
- SOR/2022-143, s. 1(F)
A.01.017 Every label of a drug for human use in dosage form shall meet the following conditions:
(a) the information that is required by these Regulations to appear on the label shall be
(i) prominently displayed on it,
(ii) readily discernible to the purchaser or consumer under the customary conditions of purchase and use, and
(iii) expressed in plain language; and
(b) the format of the label, including the manner in which its text and any graphics are displayed on it, shall not impede comprehension of the information referred to in paragraph (a).
- SOR/2014-158, s. 2
Analysts; Inspectors
A.01.020 and A.01.021 [Repealed, SOR/81-935, s. 1]
A.01.022 An inspector shall perform the functions and duties and carry out the responsibilities in respect of foods and drugs prescribed by the Act, and these Regulations.
A.01.023 The authority of an inspector extends to and includes the whole of Canada.
A.01.024 The certificate of designation required pursuant to subsection 22(2) of the Act shall
(a) certify that the person named therein is an inspector for the purpose of the Act; and
(b) be signed by
(i) the Minister and the person named in the certificate, in the case of an inspector on the staff of the Department.
(ii) [Repealed, SOR/2000-184, s. 60]
- SOR/80-500, s. 1
- SOR/92-626, s. 1
- SOR/95-548, s. 5
- SOR/2000-184, s. 60
- SOR/2018-69, s. 27
A.01.025 If authorized by a regulation made pursuant to the Broadcasting Act, inspectors shall act as representatives of the Canadian Radio-television and Telecommunications Commission for the purpose of enforcing the provisions of regulations made by the Canadian Radio-Television and Telecommunications Commission concerning the advertising of any article to which the Food and Drugs Act applies, or concerning recommendations for the prevention, treatment or cure of a disease or ailment.
A.01.026 An inspector may, for the proper administration of the Act or these Regulations, take photographs of
(a) any article that is referred to in subsection 23(2) of the Act;
(b) any place where, on reasonable grounds, he believes any article referred to in paragraph (a) is manufactured, prepared, preserved, packaged or stored; and
(c) anything that, on reasonable grounds, he believes is used or capable of being used for the manufacture, preparation, preservation, packaging or storing of any article referred to in paragraph (a).
- SOR/90-814, s. 1
Importations
A.01.040 Subject to section A.01.044, no person shall import into Canada for sale a food or drug the sale of which in Canada would constitute a violation of the Act or these Regulations.
- SOR/92-626, s. 2(F)
A.01.041 An inspector may examine and take samples of any food or drug sought to be imported into Canada.
A.01.042 If an inspector examines or takes a sample of a food or drug under section A.01.041, the inspector may submit it to an analyst for analysis or examination.
- SOR/2017-18, s. 1(E)
A.01.043 If an inspector, on examination of a sample of a food or drug or on receipt of a report of an analyst of the result of an analysis or examination of the sample, is of the opinion that the sale of the food or drug in Canada would constitute a violation of the Act or these Regulations, the inspector shall so notify in writing the collector of customs concerned and the importer.
- SOR/84-300, s. 2(E)
- SOR/2017-18, s. 2
A.01.044 (1) Where a person seeks to import a food or drug into Canada for sale and the sale would constitute a violation of the Act or these Regulations, that person may, if the sale of the food or drug would be in conformity with the Act and these Regulations after its relabelling or modification, import it into Canada on condition that
(a) the person gives to an inspector notice of the proposed importation; and
(b) the food or drug will be relabelled or modified as may be necessary to enable its sale to be lawful in Canada.
(2) No person shall sell a food or drug that has been imported into Canada under subsection (1) unless the food or drug has been relabelled or modified within three months after the importation or within such longer period as may be specified by
(a) in the case of a drug, the Minister; or
(b) in the case of food, the Minister or the President of the Canadian Food Inspection Agency.
- SOR/92-626, s. 3
- SOR/95-548, s. 5
- SOR/2000-184, s. 61
- SOR/2000-317, s. 18
- SOR/2018-69, s. 27
Exports
A.01.045 A certificate referred to in section 37 of the Act shall be signed and issued by the exporter in the form set out in Appendix III.
- SOR/80-318, s. 1
- SOR/90-814, s. 2
A.01.046 An exporter that issues a certificate referred to in paragraph 37(1)(c) of the Act in respect of a drug shall retain a copy of the certificate for five years after the day on which the drug is exported.
A.01.047 (1) For the purposes of paragraph 37(1)(d) of the Act, a drug that, by virtue of paragraph A.01.048(a), is required to be fabricated, packaged/labelled, tested, distributed or wholesaled in accordance with an establishment licence must be fabricated, packaged/labelled, tested, distributed or wholesaled by the holder of such a licence that has paid fees in connection with the licence in accordance with the Fees in Respect of Drugs and Medical Devices Order.
(2) Words and expressions used in this section have the same meaning as in subsection C.01A.001(1).
A.01.048 For the purposes of subsection 37(1.2) of the Act, the following provisions are prescribed in relation to drugs:
(a) the provisions of Division 1A of Part C;
(b) the provisions of Division 2 of Part C, except section C.02.003.2 and paragraphs C.02.009(5)(b), C.02.016(3)(b) and C.02.018(3)(c);
(c) sections C.03.001 to C.03.013; and
(d) the provisions of Division 4 of Part C, except
(i) sections C.04.003, C.04.019, C.04.020, C.04.054, C.04.055, C.04.080, C.04.082 to C.04.085 and C.04.091,
(ii) sections C.04.101 and C.04.102, paragraph C.04.117(c), sections C.04.118 to C.04.120, C.04.126 and C.04.127, paragraph C.04.128(a) and sections C.04.137, C.04.138, C.04.146, C.04.147, C.04.169, C.04.170, C.04.187 and C.04.190,
(iii) sections C.04.218 to C.04.220 and C.04.239 to C.04.241,
(iv) sections C.04.555, C.04.556, C.04.561, C.04.562, C.04.567, C.04.568, C.04.573, C.04.574, C.04.578, C.04.580, C.04.581, C.04.588, C.04.589, C.04.593, C.04.595 and C.04.596, and
(v) sections C.04.601, C.04.602, C.04.650 to C.04.656, C.04.676, C.04.677 and C.04.680 to C.04.683.
Transhipments
A.01.049 For the purposes of paragraph 38(c) of the Act, all drugs must be in bond.
Sampling
A.01.050 When taking a sample of an article under paragraph 23(2)(i) of the Act, an inspector shall inform the owner of the article or the person from whom the sample is being obtained of the inspector’s intention to submit the sample or a part of it to an analyst for analysis or examination, and
(a) where, in the opinion of the inspector, division of the procured quantity would not interfere with analysis or examination
(i) divide the quantity into three parts,
(ii) identify the three parts as the owner’s portion, the sample, and the duplicate sample and where only one part bears the label, that part shall be identified as the sample,
(iii) seal each part in such a manner that it cannot be opened without breaking the seal, and
(iv) deliver the part identified as the owner’s portion to the owner or the person from whom the sample was obtained and forward the sample and the duplicate sample to an analyst for analysis or examination; or
(b) where, in the opinion of the inspector, division of the procured quantity would interfere with analysis or examination
(i) identify the entire quantity as the sample,
(ii) seal the sample in such a manner that it cannot be opened without breaking the seal, and
(iii) forward the sample to an analyst for analysis or examination.
- SOR/90-814, s. 3
- SOR/2021-46, s. 1
A.01.051 Where the owner or the person from whom the sample was obtained objects to the procedure followed by an inspector under section A.01.050 at the time the sample was obtained, the inspector shall follow both procedures set out in that section if the owner or the person from whom the sample was obtained supplies him with a sufficient quantity of the article.
Tariff of Fees
A.01.060 The cost of analysing a sample other than for the purpose of the Act, for a department of the Government of Canada for the purpose of legal action is $15.
Labelling of Food and Drugs in Pressurized Containers
A.01.060.1 In sections A.01.061 and A.01.062,
- flame projection
flame projection means the ability of the pressurized contents of an aerosol container to ignite and the length of that ignition, when tested in accordance with official method DO-30, Determination of Flame Projection, dated October 15, 1981; (projection de flamme)
- flashback
flashback means that part of the flame projection that extends from its point of ignition back to the aerosol container when tested in accordance with official method DO-30, Determination of Flame Projection, dated October 15, 1981; (retour de flamme)
- principal display panel
principal display panel[Repealed, SOR/2000-353, s. 2]
- SOR/92-15, s. 1
- SOR/2000-353, s. 2
- SOR/2001-272, s. 6
A.01.061 (1) Subject to section A.01.063, in the case of a food or a drug packaged in a disposable metal container designed to release pressurized contents by use of a manually operated valve that forms an integral part of the container, the principal display panel of the inner and outer labels of the food or drug shall display, in accordance with sections 15 to 18 of the Consumer Chemicals and Containers Regulations, as they read on September 30, 2001, the following information:
(a) the hazard symbol set out in Column II of item 10 of Schedule II to those Regulations, accompanied by the signal word “CAUTION / ATTENTION”; and
(b) the primary hazard statement “CONTAINER MAY EXPLODE IF HEATED. / CE CONTENANT PEUT EXPLOSER S’IL EST CHAUFFÉ.”.
(2) Subject to section A.01.063, one panel of the inner and outer labels of a food or drug referred to in subsection (1) shall display, in the size required by paragraph 19(1)(b) of the Consumer Chemicals and Containers Regulations, as they read on September 30, 2001, the following additional hazard statement:
“Contents under pressure. Do not place in hot water or near radiators, stoves or other sources of heat. Do not puncture or incinerate container or store at temperatures over 50°C.
Contenu sous pression. Ne pas mettre dans l’eau chaude ni près des radiateurs, poêles ou autres sources de chaleur. Ne pas percer le contenant, ni le jeter au feu, ni le conserver à des températures dépassant 50 °C.”
(3) The requirements of subsections (1) and (2) do not apply in relation to a drug or food if the Minister determines that the design of the container, the materials used in its construction or the incorporation of a safety device eliminate its potential hazard.
- SOR/81-616, s. 1
- SOR/85-1023, s. 1
- SOR/92-15, s. 2
- SOR/2001-272, s. 7
- SOR/2018-69, s. 2
A.01.062 (1) Subject to section A.01.063, if a food or drug is packaged in a container described in subsection A.01.061(1) and has a flame projection of a length set out in column I of any of items 1 to 3 of the table to this subsection or a flashback as set out in column I of item 4 of that table, as determined by official method DO-30, Determination of Flame Projection, dated October 15, 1981, the principal display panel of the inner and outer labels of the food or drug shall display, in accordance with sections 15 to 18 of the Consumer Chemicals and Containers Regulations, as they read on September 30, 2001, the following information:
(a) the hazard symbol set out in Column II of the same item;
(b) in both official languages, the signal word set out in Column III of the same item; and
(c) in both official languages, the primary hazard statement set out in Column IV of the same item.
TABLE IS NOT DISPLAYED, SEE SOR/81-616, S. 2; SOR/92-15, S. 3
(2) In addition to the requirements of subsection (1), one panel of the inner label and outer labels of a food or drug referred to in that subsection shall display, in the size required by paragraph 19(1)(b) of the Consumer Chemicals and Containers Regulations, as they read on September 30, 2001, the following additional hazard statement:
“Do not use in presence of open flame or spark.
Ne pas utiliser en présence d’une flamme nue ou d’étincelles.”
- SOR/81-616, s. 2
- SOR/82-429, s. 1
- SOR/85-1023, s. 2
- SOR/92-15, s. 3
- SOR/2001-272, s. 8
A.01.063 (1) Where the labelled net contents of a container of a food or drug described in subsection A.01.061(1) or A.01.062(1) does not exceed 60 millilitres or 60 grams, the inner label may show only the information described in paragraph A.01.061(1)(a) or paragraphs A.01.062(1)(a) and (b), as the case may be.
(2) Where the labelled net contents of a container of a food or drug described in subsection A.01.061(1) or A.01.062(1) exceeds 60 millilitres or 60 grams but does not exceed 120 millilitres or 120 grams, the inner label may show only the information described in subsection A.01.061(1) or subsection A.01.062(1), as the case may be.
(3) Where the labelled net quantity, in a container, of a food or drug referred to in subsection A.01.061(1) or A.01.062(1) is less than 30 mL or 30 g, the hazard symbol shall be of such size as to be capable of being circumscribed by a circle with a diameter of at least 6 mm.
(4) Where a container of a food or drug, described in subsection (1) or (2) is sold in a package, the outer label may show only the information described in subsection A.01.061(2) and, where applicable, subsection A.01.062(2).
- SOR/81-616, s. 2
- SOR/92-15, s. 4
A.01.064 [Repealed, SOR/93-243, s. 2]
Security Packaging
A.01.065 (1) In this section, drug for human use means a drug that is intended for human use, whether the drug is
(a) a mouthwash;
(b) to be inhaled, ingested or inserted into the body; or
(c) for ophthalmic use.
(2) Subject to subsection (3), no person shall sell or import a drug for human use that is packaged and available to the general public in a self-service display, unless the drug is contained in a security package.
(3) Subsection (2) does not apply to lozenges.
(4) Subject to subsection (5), a statement or illustration that draws attention to the security feature of the security package referred to in subsection (2) shall be carried
(a) on the inner label of the package; and
(b) if the security feature is a part of the outer package, on the outer label.
(5) Subsection (4) does not apply if the security feature of a security package is self-evident and is an integral part of the immediate product container.
- SOR/85-141, s. 2
- SOR/88-323, s. 1
- SOR/92-664, s. 1
Exemptions
Application
A.01.066 Sections A.01.067 and A.01.068 do not apply to
(a) a drug included in Schedule I, II, III, IV or V to the Controlled Drugs and Substances Act; or
(b) a prescription drug.
- SOR/2007-288, s. 1
- SOR/2013-122, s. 2
Advertising
A.01.067 A drug is exempt from subsection 3(1) of the Act with respect to its advertisement to the general public as a preventative, but not as a treatment or cure, for any of the diseases, disorders or abnormal physical states referred to in Schedule A.1 to the Act.
- SOR/2007-288, s. 1
- SOR/2021-46, s. 10
Sale
A.01.068 A drug is exempt from subsection 3(2) of the Act with respect to its sale by a person where the drug is represented by label or is advertised by that person to the general public as a preventative, but not as a treatment or cure, for any of the diseases, disorders or abnormal physical states referred to in Schedule A.1 to the Act.
- SOR/2007-288, s. 1
- SOR/2021-46, s. 10
PART BFoods
DIVISION 1
General
B.01.001 (1) In this Part,
- agricultural chemical
agricultural chemical means any substance that is used, or represented for use, in or on a food during its production, storage or transport, and whose use results, or may reasonably be expected to result, in a residue, component or derivative of that substance in or on a food and includes any pest control product as defined in subsection 2(1) of the Pest Control Products Act, plant growth regulator, fertilizer or any adjuvant or carrier used with that substance. This definition does not include any
(a) food additive that is listed in, and used in accordance with, the tables to section B.16.100,
(b) nutritive substance that is used, recognized or commonly sold as food or as an ingredient of food,
(c) vitamin, mineral nutrient or amino acid,
(c.1) supplemental ingredient,
(d) essential oil, flavouring preparation, natural extractive, oleoresin, seasoning or spice,
(e) food packaging material or any substance of which that material is composed, or
(f) drug recommended for administration to animals that may be consumed as food; (produit chimique agricole)
- available display surface
available display surface, in respect of a prepackaged product, means
(a) the bottom of an ornamental container or the total surface area of both sides of a tag attached to the ornamental container, whichever is greater,
(b) the total surface area of both sides of a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, and
(c) the total surface area of any other package, excluding the bottom if the contents of the package leak out or are damaged when the package is turned over,
but does not include
(d) any area of a package on which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase,
(e) any part of a package that is intended to be destroyed when it is opened, other than a package of a food that is intended to be consumed by one person at a single eating occasion, or
(f) the area occupied by the universal product code; (surface exposée disponible)
- close proximity
close proximity, in respect of information that is shown on a label or a sign, means immediately adjacent to the information and without any intervening printed, written or graphic material; (à proximité)
- common name
common name means, with reference to a food,
(a) the name of the food printed in boldface type, but not in italics, in a provision of these Regulations,
(b) the name prescribed by any other regulation, or
(c) if the name of the food is not so printed or prescribed, the name by which the food is generally known or a name that is not generic and that describes the food; (nom usuel)
- Common Names for Ingredients and Components Document
Common Names for Ingredients and Components Document means the document entitled Common Names for Ingredients and Components, prepared by the Canadian Food Inspection Agency and published on its website, as amended from time to time; (document sur les noms usuels d’ingrédients et de constituants)
- component
component means an individual unit of food that is combined as an individual unit of food with one or more other individual units of food to form an ingredient; (constituant)
- daily value
daily value means, in respect of a nutrient, the quantity applicable to the nutrient according to subsection B.01.001.1(2); (valeur quotidienne)
- Directory of NFT Formats
Directory of NFT Formats means the document entitled Nutrition Labelling – Directory of Nutrition Facts Table Formats published by the Government of Canada on its website, as amended from time to time; (Répertoire des modèles de TVN)
- Directory of Nutrition Symbol Specifications
Directory of Nutrition Symbol Specifications means the document entitled Nutrition Labelling — Directory of Nutrition Symbol Specifications, published by the Government of Canada on its website, as amended from time to time; (Répertoire des spécifications des symboles nutritionnels)
- durable life
durable life means the period, commencing on the day on which a prepackaged product is packaged for retail sale, during which the product, when it is stored under conditions appropriate to that product, will retain, without any appreciable deterioration, its normal wholesomeness, palatability, nutritional value and any other qualities claimed for it by the manufacturer; (durée de conservation)
- durable life date
durable life date means the date on which the durable life of a prepackaged product ends; (date limite de conservation)
- energy value
energy value means, in respect of a food, the amount of energy made available to a person’s body when the chemical constituents of the food, including protein, fat, carbohydrate and alcohol, are metabolized following ingestion of the food by the person; (valeur énergétique)
- extended meat product
extended meat product means a meat product to which a meat product extender has been added; (produit de viande avec allongeur)
- extended poultry product
extended poultry product means a poultry product to which a poultry product extender has been added; (produit de volaille avec allongeur)
- fish product
fish product means fish or prepared fish; (produit de poisson)
- flavouring preparation
flavouring preparation includes any food for which a standard is provided in Division 10; (préparation aromatisante)
- food additive
food additive means any substance the use of which results, or may reasonably be expected to result, in it or its by-products becoming a part of or affecting the characteristics of a food, but does not include
(a) any nutritive material that is used, recognized or commonly sold as an article or ingredient of food;
(b) vitamins, mineral nutrients and amino acids, other than those listed in the tables to Division 16,
(b.1) supplemental ingredients;
(c) spices, seasonings, flavouring preparations, essential oils, oleoresins and natural extractives;
(d) agricultural chemicals, other than those listed in the tables to Division 16,
(e) food packaging materials and components thereof; and
(f) drugs recommended for administration to animals that may be consumed as food; (additif alimentaire)
- food colour
food colour means any colouring agent that is referred to in section 2 of the Marketing Authorization for Food Additives That May Be Used as Colouring Agents; (colorant alimentaire)
- fully hydrogenated
fully hydrogenated, in respect of a fat or oil, means a fat or oil that is hydrogenated and has an iodine value of 4 or less; (entièrement hydrogénée)
- functional substitute for a sweetening agent
functional substitute for a sweetening agent means, in respect of a prepackaged product, a food — other than any sweetener or sweetening agent, including any sugars — that replaces a sweetening agent and that has one or more of the functions of the sweetening agent including, sweetening, thickening, texturing or caramelizing; (substitut fonctionnel d’un agent édulcorant)
- gelling agent
gelling agent means gelatin, agar and carrageenan; (agent gélatinisant)
- ingredient
ingredient means an individual unit of food that is combined as an individual unit of food with one or more other individual units of food to form an integral unit of food that is sold as a prepackaged product; (ingrédient)
- list of cautionary statements
list of cautionary statements means the list shown on the label of a supplemented food in accordance with subsection B.29.020(1); (liste des mises en garde)
- List of Contaminants and Other Adulterating Substances in Foods
List of Contaminants and Other Adulterating Substances in Foods means the List of Contaminants and Other Adulterating Substances in Foods, published by the Department of Health on its website, as amended from time to time; (Liste de contaminants et d’autres substances adultérantes dans les aliments)
- List of Permitted Supplemental Ingredients
List of Permitted Supplemental Ingredients means the document entitled List of Permitted Supplemental Ingredients, published by the Government of Canada on its website, as amended from time to time; (Liste des ingrédients supplémentaires autorisés)
- List of Permitted Supplemented Food Categories
List of Permitted Supplemented Food Categories means the document entitled List of Permitted Supplemented Food Categories, published by the Government of Canada on its website, as amended from time to time; (Liste des catégories autorisées d’aliments supplémentés)
- main dish
main dish means a combination dish, as set out in the Table of Reference Amounts, that does not require the addition of ingredients, other than water, for its preparation and that contains food from at least two of the following categories:
(a) dairy products and their alternatives, except butter, cream, sour cream, ice cream, ice milk, sherbet and alternatives for those foods;
(b) meat products, poultry products, marine and fresh water animal products referred to in Division 21, and their alternatives such as eggs, tofu, legumes, nuts, seeds, nut or seed butters and spreads made from legumes;
(c) fruits and vegetables except pickles, relishes, olives and garnishes; and
(d) breads, breakfast cereals, rice and other grains, and alimentary pastes; (plat principal)
- marketing authorization
marketing authorization means a marketing authorization issued by the Minister under subsection 30.3(1) of the Act; (autorisation de mise en marché)
- meal replacement
meal replacement means a formulated food that, by itself, can replace one or more daily meals; (substitut de repas)
- meat product
meat product means meat, meat by-product, prepared meat or prepared meat by-product; (produit de viande)
- meat product extender
meat product extender means a food that is a source of protein and that is represented as being for the purpose of extending meat products; (allongeur de produit de viande)
- mechanically tenderized beef
mechanically tenderized beef means uncooked solid cut beef that is prepared in either of the following ways:
(a) the integrity of the surface of the beef is compromised by being pierced by blades, needles or other similar instruments; or
(b) the beef is injected with a marinade or other tenderizing solution; (boeuf attendri mécaniquement)
- monounsaturated fatty acids
monounsaturated fatty acids, monounsaturated fat, monounsaturates or monounsaturated means cis-monounsaturated fatty acids; (acides gras monoinsaturés, graisses monoinsaturées, gras monoinsaturés, lipides monoinsaturés ou monoinsaturés)
- mutiple-serving prepackaged product
mutiple-serving prepackaged product means a prepackaged product other than a single-serving prepackaged product; (produit préemballé à portions multiples)
- nutritional supplement
nutritional supplement means a food sold or represented as a supplement to a diet that may be inadequate in energy and essential nutrients. It does not include a human milk fortifier; (supplément nutritif)
- nutrition facts table
nutrition facts table means the nutrition facts table that is required by subsection B.01.401(1) to be carried on the label of a prepackaged product; (tableau de la valeur nutritive)
- nutrition symbol
nutrition symbol means a symbol that is carried on the principal display panel of a prepackaged product under subsection B.01.350(1); (symbole nutritionnel)
- omega-3 polyunsaturated fatty acids
omega-3 polyunsaturated fatty acids, omega-3 polyunsaturated fat, omega-3 polyunsaturates, omega-3 polyunsaturated or omega-3 means
(a) 9-cis, 12-cis, 15-cis octadecatrienoic acid or α-linolenic acid,
(b) 8-cis, 11-cis, 14-cis, 17-cis eicosatetraenoic acid,
(c) 5-cis, 8-cis, 11-cis, 14-cis, 17-cis eicosapentaenoic acid or EPA,
(d) 7-cis, 10-cis, 13-cis, 16-cis, 19-cis docosapentaenoic acid, or
(e) 4-cis, 7-cis, 10-cis, 13-cis, 16-cis, 19-cis docosahexaenoic acid or DHA; (acides gras polyinsaturés oméga-3, graisses polyinsaturées oméga-3, gras polyinsaturés oméga-3, lipides polyinsaturés oméga-3, polyinsaturés oméga-3 ou oméga-3)
- omega-6 polyunsaturated fatty acids
omega-6 polyunsaturated fatty acids, omega-6 polyunsaturated fat, omega-6 polyunsaturates, omega-6 polyunsaturated or omega-6 means
(a) 9-cis, 12-cis octadecadienoic acid or linoleic acid,
(b) 6-cis, 9-cis, 12-cis octadecatrienoic acid,
(c) 8-cis, 11-cis, 14-cis eicosatrienoic acid or di-homo-γ-linolenic acid,
(d) 5-cis, 8-cis, 11-cis, 14-cis eicosatetraenoic acid or arachidonic acid,
(e) 7-cis, 10-cis, 13-cis, 16-cis docosatetraenoic acid, or
(f) 4-cis, 7-cis, 10-cis, 13-cis, 16-cis docosapentaenoic acid; (acides gras polyinsaturés oméga-6, graisses polyinsaturées oméga-6, gras polyinsaturés oméga-6, lipides polyinsaturés oméga-6, polyinsaturés oméga-6 ou oméga-6)
- ornamental container
ornamental container means a container that, except on the bottom, does not have any promotional or advertising material thereon, other than a trademark or common name and that, because of any design appearing on its surface or because of its shape or texture, appears to be a decorative ornament and is sold as a decorative ornament in addition to being sold as the container of a product; (emballage décoratif)
- overage
overage means the amount of a vitamin or mineral nutrient that is, within the limits of good manufacturing practice, added to a food in excess of the amount declared on the label, in order to ensure that the amount of the vitamin or mineral nutrient declared on the label is maintained throughout the durable life of the food; (surtitrage)
- parts per million
parts per million[Repealed, SOR/2010-94, s. 1]
- parts per million
parts per million or p.p.m. means parts per million by weight unless otherwise stated; (parties par million ou p.p.m.)
- per cent
per cent or % means per cent by weight, unless otherwise stated; (pour cent ou %)
- point
point means the unit of measurement for type size that is known as a PostScript point and is equal to 0.3527777778 mm; (point)
- polyunsaturated fatty acids
polyunsaturated fatty acids, polyunsaturated fat, polyunsaturates or polyunsaturated means cis-methylene interrupted polyunsaturated fatty acids; (acides gras polyinsaturés, graisses polyinsaturées, gras polyinsaturés, lipides polyinsaturés ou polyinsaturés)
- poultry product
poultry product means poultry meat, prepared poultry meat, poultry meat by-product or prepared poultry meat by-product; (produit de volaille)
- poultry product extender
poultry product extender means a food that is a source of protein and that is represented as being for the purpose of extending poultry products; (allongeur de produit de volaille)
- prepackaged meal
prepackaged meal means a prepackaged selection of foods for one individual that requires no preparation other than heating and that contains at least one serving, as described in Canada’s Food Guide to Healthy Eating, published in 1992 by the Department of Supply and Services by authority of the Minister of National Health and Welfare, of
(a) meat, fish, poultry, legumes, nuts, seeds, eggs or milk or milk products other than butter, cream, sour cream, ice-cream, ice milk and sherbet; and
(b) vegetables, fruit or grain products; (repas préemballé)
- prepackaged product
prepackaged product means any food that is contained in a package in the manner in which it is ordinarily sold to or used or purchased by a person; (produit préemballé)
- principal display panel
principal display panel means, despite the meaning assigned to that term in section A.01.010,
(a) in the case of a label that is applied to a consumer prepackaged food within the meaning of section 1 of the Safe Food for Canadians Regulations, the principal display panel as described in paragraphs (a) to (c) of the definition of that term in that section,
(b) in the case of a label that is applied to a prepackaged product other than a consumer prepackaged food subject to the Safe Food for Canadians Regulations, the part of the label that is applied to all or part of any side or surface of the container that is displayed or visible under normal or customary conditions of sale or use and, if the container does not have such a side or surface, the part of the label that is applied to any part of the container except on the bottom, or
(c) in the case of a label that is applied to a food that is not a prepackaged product, the part of the label that is applied to all or part of the side or surface of the food that is displayed or visible under normal or customary conditions of sale or use; (espace principal)
- principal display surface
principal display surface, in respect of a prepackaged product, means
(a) if the package has a surface that is displayed or visible under customary conditions of sale or use, the total area of that surface, excluding any surface that is the top of the package,
(b) if the package has a lid that is the part of the package that is displayed or visible under customary conditions of sale or use, the total area of the top surface of the lid,
(c) if the package does not have a particular surface that is displayed or visible under customary conditions of sale or use, 40% of the total surface area of the package, excluding any surface area that is its top and bottom, if it is possible for that proportion of the total surface area to be displayed or visible under customary conditions of sale or use,
(d) if the package is a bag with surfaces of equal dimensions, the total area of one of the surfaces,
(e) if the package is a bag with surfaces of different dimensions, the total area of one of the largest surfaces,
(f) despite paragraphs (a) to (e), if the package does not have a surface that is displayed or visible under customary conditions of sale or use to which a label can be applied, the total area of one side of a tag that is attached to the package,
(g) despite paragraphs (a) to (e), if the package contains wine that is exposed for sale, any part of the surface of the package, excluding its top and bottom, that can be seen without having to turn the package, and
(h) if the package is a wrapper or confining band that is so narrow in relation to the size of the food that it cannot reasonably be considered to have any surface that is displayed or visible under customary conditions of sale or use, the total area of one side of a tag that is attached to the package; (principale surface exposée)
- reasonable daily intake
reasonable daily intake, in respect of a food set out in Column I of an item of Schedule K, means the amount of that food set out in Column II of that item; (ration quotidienne raisonnable)
- recommended daily intake
recommended daily intake[Repealed, SOR/2016-305, s. 1]
- reference amount
reference amount means, in respect of a food set out in column 1 of the Table of Reference Amounts, the amount of that food set out in column 2; (quantité de référence)
- reference standard
reference standard[Repealed, SOR/2016-305, s. 1]
- saturated fatty acids
saturated fatty acids, saturated fat, saturates or saturated means all fatty acids that contain no double bonds; (acides gras saturés, graisses saturées, gras saturés, lipides saturés ou saturés)
- simulated meat product
simulated meat product means any food that does not contain any meat product, poultry product or fish product but that has the appearance of a meat product; (simili-produit de viande)
- simulated poultry product
simulated poultry product means any food that does not contain any poultry product, meat product or fish product but that has the appearance of a poultry product; (simili-produit de volaille)
- single-serving prepackaged product
single-serving prepackaged product means a prepackaged product in respect of which the net quantity of food in the package is the serving of stated size for the food as set out in paragraph B.01.002A(1)(b); (produit préemballé à portion individuelle)
- sugars
sugars means all monosaccharides and disaccharides; (sucres)
- sugars-based ingredient
sugars-based ingredient means, in respect of a prepackaged product,
(a) an ingredient that is a monosaccharide or disaccharide or a combination of these;
(b) an ingredient that is a sweetening agent other than one referred to in paragraph (a); and
(c) any other ingredient that contains one or more sugars and that is added to the product as a functional substitute for a sweetening agent; (ingrédient à base de sucres)
- supplemental ingredient
supplemental ingredient means a nutrient — including a vitamin, mineral nutrient or amino acid — or any other substance listed in column 1 of the List of Permitted Supplemental Ingredients and added as an ingredient to a food in accordance with the applicable conditions of use set out in columns 2 to 5; (ingrédient supplémentaire)
- supplemented food
supplemented food means a prepackaged product that belongs to a food category set out in column 1 of the List of Permitted Supplemented Food Categories and to which a supplemental ingredient has been added, but does not include
(a) a food for special dietary use as defined in section B.24.001 and referred to in any of paragraphs B.24.003(1)(f) to (f.2) and (h) to (j), even if the food for special dietary use is also a gluten-free food referred to in paragraph B.24.003(1)(g);
(b) a food that is labelled or advertised for consumption by
(i) infants as defined in section B.25.001,
(ii) children one year of age or older but less than four years of age, or
(iii) women who are pregnant or breastfeeding;
(c) any of the following foods set out in column I of the Table to section D.03.002:
(i) a food referred to in any of items 1, 2.1, 2.2, 4, 5, 7, 8, 9.1, 10 to 13, 15, 17 to 19, 21 to 25 and 27, and
(ii) prepackaged ice;
(d) a food that has not been processed or that has been minimally processed; or
(e) a beverage with an alcohol content of more than 0.5%; (aliment supplémenté)
- supplemented food caution identifier
supplemented food caution identifier means the identifier carried on the principal display panel of a supplemented food under subsection B.29.021(1); (identifiant des aliments supplémentés avec mise en garde)
- supplemented food facts table
supplemented food facts table means the supplemented food facts table required by subsection B.29.002(1) to be carried on the label of a supplemented food; (tableau des renseignements sur les aliments supplémentés)
- sweetener
sweetener means any sweetener that is referred to in section 2 of the Marketing Authorization for Food Additives That May Be Used as Sweeteners; (édulcorant)
- sweetening agent
sweetening agent includes any food for which a standard is provided in Division 18, but does not include those food additives listed in the tables to Division 16; (agent édulcorant)
- Table of Daily Values
Table of Daily Values means the document entitled Nutrition Labelling – Table of Daily Values published by the Government of Canada on its website, as amended from time to time; (Tableau des valeurs quotidiennes)
- Table of Permitted Nutrient Content Statements and Claims
Table of Permitted Nutrient Content Statements and Claims means the document entitled Nutrition Labelling — Table of Permitted Nutrient Content Statements and Claims, published by the Government of Canada on its website, as amended from time to time; (Tableau des mentions et des allégations autorisées concernant la teneur nutritive)
- Table of Reference Amounts
Table of Reference Amounts means the document entitled Nutrition Labelling – Table of Reference Amounts for Food published by the Government of Canada on its website, as amended from time to time; (Tableau des quantités de référence)
- trans fatty acids
trans fatty acids, trans fat or trans means unsaturated fatty acids that contain one or more isolated or non-conjugated double bonds in a trans-configuration; (acides gras trans, graisses trans, gras trans, lipides trans ou trans)
- unstandardized food
unstandardized food means any food for which a standard is not prescribed in this Part; (aliment non normalisé)
- weighted recommended nutrient intake
weighted recommended nutrient intake, in respect of a vitamin or mineral nutrient set out in column I of Table II to Division 1 of Part D or in column I of Table II to Division 2 of Part D, means the amount set out in column III; (apport nutritionnel recommandé pondéré)
- yolk-replaced egg
yolk-replaced egg means a food that
(a) does not contain egg yolk but contains fluid, dried or frozen egg albumen or mixtures thereof,
(b) is intended as a substitute for whole egg, and
(c) meets the requirements of section B.22.032; (oeuf à jaune substitué)
(2) The definitions in this subsection apply for the purposes of the Act.
- agricultural chemical
agricultural chemical has the same meaning as in subsection (1). (produit chimique agricole)
- food additive
food additive has the same meaning as in subsection (1). (additif alimentaire)
(3) The following definitions apply in this Division.
- human milk fortifier
human milk fortifier has the same meaning as in section B.25.001. (fortifiant pour lait humain)
- human milk substitute
human milk substitute has the same meaning as in section B.25.001. (succédané de lait humain)
(4) For the purposes of the definitions supplemental ingredient and supplemented food in subsection (1), if a supplemented food is used as an ingredient in the manufacture of a second supplemented food, a supplemental ingredient in the first supplemented food is deemed to also have been added as a supplemental ingredient to — and not to be a component of an ingredient of — the second supplemented food if it is contained in the second supplemented food in accordance with the applicable conditions of use set out in columns 2 to 5 of the List of Permitted Supplemental Ingredients.
- SOR/78-403, s. 1(F)
- SOR/79-23, s. 1
- SOR/81-83, s. 1
- SOR/81-617, s. 1
- SOR/88-336, s. 1
- SOR/88-559, s. 1
- SOR/89-175, s. 1
- SOR/91-124, s. 1
- SOR/91-527, s. 1
- SOR/93-276, s. 1
- SOR/95-474, s. 1
- SOR/98-580, s. 1(F)
- SOR/2000-353, s. 3
- SOR/2003-11, s. 1
- err.(E), Vol. 137, No. 5
- SOR/2005-98, s. 1
- SOR/2008-181, s. 1
- SOR/2008-182, s. 1
- SOR/2010-94, s. 1
- SOR/2011-278, s. 1
- 2014, c. 20, s. 366(E)
- SOR/2014-99, s. 1
- SOR/2016-74, s. 1
- SOR/2016-305, s. 1
- SOR/2018-108, s. 393
- SOR/2021-57, s. 1
- SOR/2022-143, s. 2
- SOR/2022-168, s. 1
- SOR/2022-169, s. 1
B.01.001.1 (1) For the purpose of subsection (2), the term fat used in the Table of Daily Values means all fatty acids expressed as triglycerides.
(2) The daily value for a nutrient in a food is
(a) in the case of a nutrient set out in column 1 of Part 1 of the Table of Daily Values, the quantity
(i) set out in column 2, if the food is intended solely for children one year of age or older but less than four years of age, and
(ii) set out in column 3, if the food is intended for children one year of age or older but less than four years of age or for children four years of age or older and adults; and
(b) in the case of a vitamin or mineral nutrient set out in column 1 of Part 2 of the Table of Daily Values, the quantity
(i) set out in column 2, if the food is intended solely for infants six months of age or older but less than one year of age,
(ii) set out in column 3, if the food is intended for infants six months of age or older but less than one year of age or for children one year of age or older but less than four years of age, and
(iii) set out in column 4, in any other case.
(3) Subsection (2) does not apply if the food is
(a) a human milk fortifier; or
(b) a human milk substitute intended solely for infants less than six months of age.
- SOR/2003-11, s. 2
- SOR/2016-305, s. 2
- SOR/2021-57, s. 2
B.01.002 Each provision in this Part in which the symbol [S] appears between the provision number and the name of the food described in that provision prescribes the standard of composition, strength, potency, purity, quality or other property of that food and a provision in which the symbol does not appear does not prescribe a standard for a food.
- SOR/79-752, s. 1
B.01.002A (1) For the purposes of this Part, a serving of stated size of a food shall be
(a) based on the food as offered for sale;
(b) in either of the following cases, the net quantity of the food in the package:
(i) if the quantity of food in the package can reasonably be consumed by one person at a single eating occasion, or
(ii) if the package contains less than 200% of the reference amount for the food; and
(c) in all other cases, the amount indicated for the food according to the criteria set out in column 3A of the Table of Reference Amounts.
(2) A serving of stated size of a food shall be expressed as follows:
(a) in the case of a single-serving prepackaged product to which paragraph (1)(b) applies, per package and using the following units:
(i) in grams, if the net quantity of the food is shown on the label by weight or by count, and
(ii) in millilitres, if the net quantity of the food is shown on the label by volume; and
(b) in the case of a multiple-serving prepackaged product to which paragraph (1)(c) applies, according to the following units set out in column 3B of the Table of Reference Amounts and according to the manner set out in that column:
(i) the household measure that applies to the product, and
(ii) the metric measure that applies to the product.
- SOR/88-559, s. 2
- SOR/2003-11, s. 3
- SOR/2016-305, s. 3
B.01.003 (1) The following foods shall carry a label when offered for sale:
(a) all prepackaged products other than
(i) prepackaged confections, commonly known as one bite confections, that are sold individually, and
(ii) prepackaged products consisting of fresh fruits or fresh vegetables that are packaged in a wrapper or confining band of less than 1/2 inch in width;
(b) meat and meat by-products that are barbecued, roasted or broiled on the retail premises;
(c) poultry, poultry meat or poultry meat by-products that are barbecued, roasted or broiled on the retail premises;
(d) horse-meat or horse-meat by-product;
(e) any substance or mixture of substances for use as a food additive or food additive preparation; and
(f) flour and whole wheat flour that has been treated with gamma radiation from Cobalt 60 Source.
(2) [Repealed, SOR/79-23, s. 2]
- SOR/79-23, s. 2
B.01.004 (1) All or part of the label referred to in section B.01.003 shall be applied
(a) in the case of a prepackaged product, to the container in which the prepackaged product is sold; and
(b) in the case of a food that is not a prepackaged product, to the food itself.
(2) The label shall be applied in such a manner that the container of the prepackaged product or the food, as the case may be, will bear the label at the time it is sold.
- SOR/84-300, s. 3
B.01.005 (1) Subject to subsections (2) to (5), the information required to be shown on a label shall not be shown on that part of the label, if any, that is applied to the bottom of a food or container.
(2) The information required to be shown on a label may be shown on that part of the label, if any, that is applied to the bottom of a food or to the bottom of a container if such information is also shown in those parts of the label that are not applied to the bottom of the food or container.
(3) Despite subsection (2), if the container of a prepackaged product is an ornamental container and the label is applied to the bottom of the container, the information required to be shown may be shown on the label that is applied to the bottom of the container.
(4) Despite subsection (2), the information required by subparagraph B.01.007(1.1)(b)(i) and paragraphs B.24.103(g), B.24.202(d), B.24.304(h), B.25.020(1)(h) and B.25.057(1)(f) and (2)(f) may be shown on that part of the label that is applied to the bottom of the package if a reference to where that information is located on the label appears elsewhere on the label.
(4.1) Despite subsection (2), the information required by paragraph B.27.005(a) may be shown on that part of the label that is applied to the bottom of the package.
(5) Despite subsection (2), the nutrition facts table, supplemented food facts table and list of cautionary statements may be shown on that part of the label that is applied to the bottom of the food or container if the available display surface includes the bottom.
- SOR/79-529, s. 1
- SOR/92-626, s. 4
- SOR/2003-11, s. 4
- SOR/2021-57, s. 3
- SOR/2022-143, s. 3
- SOR/2022-169, s. 2
B.01.006 (1) The common name of the food shall be shown on the principal display panel.
(2) Notwithstanding subsection (1), the common name of a fresh fruit or fresh vegetable that is prepackaged in such a manner that the fruit or vegetable is visible and identifiable in the package is not required to be shown on the label.
- SOR/79-23, s. 3
- SOR/92-626, s. 5
B.01.006.1 Except as otherwise provided in these Regulations or in the Safe Food for Canadians Regulations, if a prepackaged product is likely to be mistaken for another food, words describing the true nature of the prepackaged product so as to distinguish it from the other food shall appear on the principal display panel and may be in regard to
(a) its type of liquid packaging medium, including brine, vegetable oil or syrup;
(b) its style or form, including firm, extra firm, whole, sliced or diced; and
(c) its condition, including dried, concentrated, reconstituted, carbonated or smoked, which, if shown, shall form part of the common name.
B.01.007 (1) In this section, packaging date means
(a) the date on which a food is placed for the first time in a package in which it will be offered for sale to a consumer; or
(b) the date on which a prepackaged product is weighed by a retailer in a package in which it will be offered for sale for the first time to a consumer.
(1.1) The following information shall be shown on any part of the label:
(a) the identity and principal place of business of the person by or for whom the food was manufactured or produced;
(b) where a prepackaged product having a durable life of 90 days or less is packaged at a place other than the retail premises from which it is to be sold,
(i) the durable life date, and
(ii) instructions for the proper storage of the prepackaged product if it requires storage conditions that differ from normal room temperature; and
(c) where a prepackaged product having a durable life of 90 days or less is packaged on the retail premises from which it is to be sold,
(i) the packaging date, and
(ii) the durable life of the food, except when the durable life appears on a poster next to the food.
(1.2) The packaging date referred to in paragraph (1.1)(c) shall be shown in the form and manner prescribed for the durable life date by subsections (4) and (5) and the terms “best before” and “meilleur avant” on the label shall be replaced by the terms “packaged on” and “empaqueté le”.
(2) Paragraph (1.1)(a) does not apply to fresh fruits or fresh vegetables that are prepackaged on retail premises in such a manner that the fruits or vegetables are visible and identifiable in the package.
(3) Paragraphs (1.1)(b) and (c) do not apply to
(a) prepackaged products consisting of fresh fruits or fresh vegetables;
(b) prepackaged individual portions of food that are served by a restaurant or other commercial enterprise with meals or snacks;
(c) prepackaged individual servings of food that are prepared by a commissary and sold by automatic vending machines or mobile canteens; or
(d) prepackaged donuts.
(4) The durable life date shall be shown in the following manner:
(a) the words “best before” and “meilleur avant” shall be shown grouped together with the durable life date unless a clear explanation of the significance of the durable life date appears elsewhere on the label;
(b) where, for the sake of clarity, it is necessary to show the year in which the durable life date occurs, the year shall be shown first and shall be expressed by at least the last two numbers of the year;
(c) the month shall be shown in words after the year, if the year is shown, and may be abbreviated as prescribed by subsection (5); and
(d) the day of the month shall be shown after the month and shall be expressed in numbers.
(5) The month of the durable life date, when abbreviated, shall be abbreviated as follows and only one such abbreviation shall be used for the English language and the French language:
- JAfor JANUARY
- FEfor FEBRUARY
- MRfor MARCH
- ALfor APRIL
- MAfor MAY
- JNfor JUNE
- JLfor JULY
- AUfor AUGUST
- SEfor SEPTEMBER
- OCfor OCTOBER
- NOfor NOVEMBER
- DEfor DECEMBER
(6) Except as otherwise provided in these Regulations, no person shall use a durable life date marking system on the label of a prepackaged product or in advertising a prepackaged product other than the marking system set out in this section.
(7) Paragraph (1.1)(b) does not apply to prepackaged fresh yeast, if
(a) the date on which it is estimated that the product has lost its effectiveness is shown on the label in the form and manner prescribed for the durable life date by subsections (4) and (5); and
(b) the terms “best before” and “meilleur avant” are replaced by the terms “use by” and “employez avant”.
- SOR/79-23, s. 4
- SOR/79-529, s. 2
- SOR/88-291, s. 1
- SOR/92-626, s. 6
B.01.008 (1) The following information shall be shown grouped together on any part of the label:
(a) any information required by these Regulations, other than
(i) the information required to appear on the principal display panel, nutrition facts table or supplemented food facts table,
(ii) the information required by subsection B.01.005(4), sections B.01.007, B.01.301, B.01.305, B.01.311, B.01.503, B.01.513 and B.01.601, paragraphs B.24.103(g), B.24.202(d), B.24.304(h), B.25.020(1)(e), (f) and (h), B.25.057(1)(f) and (2)(f), B.27.005(a) and sections B.29.020 and B.29.026, and
(iii) any statement required to be shown on the label of a supplemented food in accordance with column 5 of the List of Permitted Supplemental Ingredients; and
(b) where a prepackaged product consists of more than one ingredient, a list of all ingredients, including, subject to section B.01.009, components, if any.
(2) Paragraph (1)(b) does not apply to
(a) prepackaged products packaged from bulk on retail premises, except prepackaged products that are a mixture of nuts;
(b) prepackaged individual portions of food that are intended solely to be served by a restaurant or other commercial enterprise with meals or snacks;
(c) prepackaged individual servings of food that are prepared by a commissary and sold by automatic vending machines or mobile canteens;
(d) prepackaged meat and meat by-products that are barbecued, roasted or broiled on the retail premises;
(e) prepackaged poultry, poultry meat or poultry meat by-products that are barbecued, roasted or broiled on the retail premises;
(f) Bourbon whisky and prepackaged products subject to compositional standards in Division 2; or
(g) prepackaged products subject to compositional standards in Division 19.
(3) Despite paragraph (1)(b), the following items are not required to be shown on the label of a prepackaged product:
(a) wax coating compounds and their components, used as an ingredient or component of prepackaged fresh fruit or vegetables;
(b) sausage casings, used as an ingredient or component of prepackaged sausages;
(c) hydrogen, used as an ingredient or component of a prepackaged product if used for hydrogenation purposes;
(d) components of ingredients of a sandwich, if the sandwich is made with bread; and
(e) added water used as an ingredient that has been removed during the manufacture of the prepackaged product.
(4) to (10) [Repealed, SOR/2016-305, s. 4]
- SOR/79-23, s. 5
- SOR/88-559, s. 3
- SOR/92-626, s. 7
- SOR/93-145, s. 1
- SOR/2003-11, s. 5
- SOR/2011-28, s. 1
- SOR/2016-305, s. 4
- SOR/2021-57, s. 4
- SOR/2022-143, s. 5
- SOR/2022-169, s. 3
B.01.008.1 (1) Information appearing on the label of a prepackaged product according to sections B.01.008.2 to B.01.010.4, B.01.014 and B.29.020 shall be shown
(a) in a single colour of type that is a visual equivalent of 100% solid black type on a white background or a uniform neutral background with a maximum 5% tint of colour;
(b) in a single standard sans serif font that is not decorative and in a manner that the characters never touch each other or any differentiating feature under subsection B.01.008.2(2);
(c) in type of normal or condensed width that is not scaled down so that the characters take up less space horizontally and, if a nutrition facts table or supplemented food facts table appears on the label, the width of type must be the same as that required for the type used to show the nutrients that appear in the nutrition facts table or the supplemental ingredients that appear in the supplemented food facts table, as the case may be;
(d) in regular type, except as otherwise provided in those sections; and
(e) in type that is the same height that is not less than 1.1 mm with identical leading of not less than 2.5 mm, except as otherwise provided in this section or those sections.
(2) Despite paragraph (1)(a), a list of ingredients appearing on the label of the following prepackaged products is not required to be shown on a white background or a uniform neutral background with a maximum 5% tint of colour:
(a) a prepackaged product that is intended solely for use as an ingredient in the manufacture of other prepackaged products intended for sale to a consumer at the retail level or as an ingredient in the preparation of food by a commercial or industrial enterprise or an institution; and
(b) a prepackaged product that is a ready-to-serve multiple-serving prepackaged product intended solely to be served in a commercial or industrial enterprise or an institution.
(3) Except as otherwise provided in subsection (4) and sections B.01.008.2 to B.01.010.4, if a nutrition facts table or supplemented food facts table appears on the label of a prepackaged product and the type size of the nutrients shown in the nutrition facts table or the supplemental ingredients shown in the supplemented food facts table is not less than 8 points, the information referred to in subsection (1) must be shown in type that is
(a) the same height as the type in which the nutrients are shown in the nutrition facts table or the supplemental ingredients are shown in the supplemented food facts table, as the case may be; and
(b) of a height that is not less than 1.4 mm with identical leading of not less than 3.2 mm.
(4) A title that introduces a list of ingredients, a food allergen source, gluten source and added sulphites statement as defined in subsection B.01.010.1(1), a declaration referred to in subsection B.01.010.4(1) or a list of cautionary statements may be shown in type that is of a greater height than the type used to show the information in either of the lists, the statement or the declaration, as the case may be.
(5) If more than one of the titles referred to in subsection (4) appears on a label, the characters of each title must be of the same height.
(6) For the purpose of this section, the height of type means the height of the lower case letter “x”.
- SOR/2016-305, s. 5
- SOR/2022-168, s. 2
- SOR/2022-169, s. 4
B.01.008.2 (1) A list of ingredients shall be introduced by a title that shall
(a) consist of the term
(i) “Ingredients”, “Ingredients:” or “Ingredients :” in the English version of the list, and
(ii) “Ingrédients”, “Ingrédients:” or “Ingrédients:” in the French version of the list;
(b) be shown in bold type; and
(c) be shown without any intervening printed, written or graphic material appearing between the title and the first ingredient shown in the list.
(2) A list of ingredients shall be shown in a manner that clearly differentiates it on the label, by means of one or both of
(a) graphics in the form of a solid-line border around the list or one or more solid lines appearing immediately above, below or at the sides of the list that are the same colour as that of the type used to show the information referred to in subsection B.01.008.1(1); and
(b) a background colour that creates a contrast between the background colour of the list and the background colour used on the adjacent surface of the label, other than the surface used to display
(i) a food allergen source, gluten source and added sulphites statement as defined in subsection B.01.010.1(1),
(ii) a declaration referred to in subsection B.01.010.4(1),
(iii) any statement referred to in subsection B.01.014(1),
(iv) a nutrition facts table,
(v) a supplemented food facts table, or
(vi) a list of cautionary statements.
(3) In a list of ingredients, ingredients shall be shown
(a) in descending order of their proportion by weight of the prepackaged product, determined before they are combined to form the prepackaged product;
(b) in lower case letters, except that upper case letters shall be used to show
(i) the first letter of each ingredient or, in the case of a food additive or supplemental ingredient shown in whole or in part by an acronym, the entire acronym, and
(ii) the alpha-descriptor that forms a part of the common name for a food additive, vitamin, supplemental ingredient or microorganism; and
(c) separated by a bullet point or a comma.
(4) Despite paragraph (3)(a), the following ingredients may be shown at the end of the list of ingredients, in any order:
(a) spices, herbs and other seasonings, other than salt added separately, if the total weight of those other seasonings is no more than two per cent of the total weight of ingredients used in the manufacture of the prepackaged product excluding added water used as an ingredient that has been removed during its manufacture;
(b) natural flavours and artificial flavours;
(c) flavour enhancers;
(d) food additives, except ingredients of food additive preparations or mixtures of substances for use as a food additive;
(e) vitamins;
(f) salts or derivatives of vitamins;
(g) mineral nutrients;
(h) salts of mineral nutrients; and
(i) supplemental ingredients.
(5) In a list of ingredients, the components of an ingredient shall be shown
(a) in parentheses, immediately after the ingredient, unless the source of a food allergen or gluten is shown immediately after the ingredient, in which case components of the ingredient shall be shown immediately after that source;
(b) in descending order of their proportion by weight of the ingredient, determined before the components are combined to form the ingredient;
(c) in lower case letters, except that upper case letters shall be used to show
(i) in the case of a food additive or supplemental ingredient shown in whole or in part by an acronym, the entire acronym, and
(ii) the alpha-descriptor that forms a part of the common name for a food additive, vitamin, supplemental ingredient or microorganism; and
(d) separated by a comma.
(6) Despite paragraph B.01.008(1)(b) and paragraphs (5)(a) and (b), but subject to section B.01.009, if one or more components of an ingredient are required by these Regulations to be shown in a list of ingredients, the ingredient is not required to be shown in the list if all components of the ingredient are shown in the list by their common names and in accordance with subsection (3) as if they were ingredients.
(7) In a list of ingredients, the source of a food allergen or gluten shall be shown
(a) immediately after the ingredient or component to which it applies in accordance with subsections B.01.010.1(8) and (10);
(b) entirely in lower case letters; and
(c) separated by a comma from any other source of a food allergen or gluten that is shown for the same ingredient or component.
(8) If the English and French versions of a list of ingredients appear on the label, they shall be displayed on a continuous surface of the available display surface, but need not be on the same continuous surface of the available display surface.
(9) If the English and French versions of a list of ingredients appear on the same continuous surface of the label, the version that follows the other version shall not begin on the same line as that on which the other version ends, except in the case of a prepackaged product that has an available display surface of less than 100 cm2.
B.01.008.3 (1) If a prepackaged product contains one or more sugars-based ingredients, despite the order of presentation referred to in paragraph B.01.008.2(3)(a), the sugars-based ingredient or ingredients shall be shown in the list of ingredients, in parentheses, immediately following the term
(a) “Sugars” in the English version of the list; and
(b) “Sucres” in the French version of the list.
(2) The term “Sugars” referred to in subsection (1) shall be shown in the list of ingredients
(a) in descending order of the proportion by weight of all the sugars-based ingredients in the prepackaged product, before they are combined to form the product; and
(b) separated from other ingredients by a bullet point or a comma.
(3) Each sugars-based ingredient mentioned immediately following the term “Sugars” shall be shown,
(a) despite paragraph B.01.008.2(3)(b), entirely in lower case letters; and
(b) in the case of more than one sugars-based ingredient,
(i) in descending order of its proportion by weight of the prepackaged product as prescribed by subsection B.01.008.2(3), and
(ii) separated by a comma.
(4) Subsections (1) to (3) do not apply to the following prepackaged products:
(a) a sweetening agent;
(b) fruit or vegetable juice or vegetable drink that does not contain any sweetening agent, as well as any mixture of those juices and drinks, including any of those juices and drinks
(i) to which fruit or vegetable purée or any mixture of those purées has been added,
(ii) that are reconstituted, or
(iii) that are a concentrate intended for dilution and consumption as juice or drink;
(c) fruit or vegetable purée that does not contain any sweetening agent as well as any mixture of those purées;
(d) prepackaged products that contain only one sugars-based ingredient that contains the word “sugar” in its common name;
(e) formulated liquid diets, human milk fortifiers and human milk substitutes; and
(f) prepackaged products that contain less than 0.5 g of sugars per serving of stated size.
- SOR/2016-305, s. 5
- SOR/2021-57, s. 5
- SOR/2022-143, s. 7
- SOR/2022-197, s. 4
B.01.009 (1) Components of ingredients or of classes of ingredients set out in the following table are not required to be shown on a label:
TABLE
Item Ingredient 1 butter 2 margarine 3 shortening 4 lard 5 leaf lard 6 monoglycerides 7 diglycerides 8 rice 9 starches or modified starches 10 breads subject to compositional standards in sections B.13.021 to B.13.029 11 flour 12 soy flour 13 graham flour 14 whole wheat flour 15 baking powder 16 milks subject to compositional standards in sections B.08.003 to B.08.027 17 chewing gumbase 18 sweetening agents subject to compositional standards in sections B.18.001 to B.18.018 19 cocoa, low-fat cocoa 20 salt 21 vinegars subject to compositional standards in sections B.19.003 to B.19.007 22 Bourbon whisky and alcoholic beverages subject to compositional standards in sections B.02.001 to B.02.134 23 cheese for which a standard is prescribed in Division 8, if the total amount of cheese in a prepackaged product is less than 10 per cent of that packaged product 24 jams, marmalades and jellies subject to compositional standards in sections B.11.201 to B.11.241 when the total amount of those ingredients is less than 5 per cent of a prepackaged product 25 olives, pickles, relish and horse-radish when the total amount of those ingredients is less than 10 per cent of a prepackaged product 26 vegetable or animal fats or oils for which a standard is prescribed in Division 9, and modified, interesterified or fully hydrogenated vegetable or animal fats or oils, if the total quantity of those fats and oils that are contained in a prepackaged product is less than 15 per cent of the prepackaged product 27 prepared or preserved meat, fish, poultry meat, meat by-product or poultry by-product when the total amount of those ingredients is less than 10 per cent of a prepackaged product that consists of an unstandardized food 28 alimentary paste that does not contain egg in any form or any flour other than wheat flour 29 bacterial culture 30 hydrolyzed plant protein 31 carbonated water 32 whey, whey powder, concentrated whey, whey butter and whey butter oil 33 mould culture 34 chlorinated water and fluorinated water 35 gelatin 36 toasted wheat crumbs used in or as a binder, filler or breading in or on a food product (2) Subject to subsection (3), where a preparation or mixture set out in the table to this subsection is added to a food, the ingredients and components of the preparation or mixture are not required to be shown on the label of that food.
TABLE
Item Preparation/Mixture 1 food colour preparations 2 flavouring preparations 3 artificial flavouring preparations 4 spice or herb mixtures 5 seasoning mixtures, other than those set out in item 4 and salt added separately, if the total weight of those seasoning ingredients is no more than 2% of the total weight of ingredients used in the manufacture of the prepackaged product excluding added water used as an ingredient that has been removed during its manufacture 6 vitamin preparations 7 mineral preparations 8 food additive preparations 9 rennet preparations 10 food flavour-enhancer preparations 11 compressed, dry, active or instant yeast preparations (3) Where a preparation or mixture set out in the table to subsection (2) is added to a food, and the preparation or mixture contains one or more of the following ingredients or components, those ingredients or components shall be shown by their common names in the list of the ingredients of the food to which they are added as if they were ingredients of that food:
(a) salt;
(b) glutamic acid or its salts;
(c) hydrolyzed plant protein;
(d) aspartame;
(e) potassium chloride;
(f) any ingredient or component that performs a function in, or has any effect on, that food; and
(g) any supplemental ingredient.
(4) Notwithstanding subsections (1) and (2), where any of the following components is contained in an ingredient set out in the tables to those subsections, that component shall be shown in the list of ingredients:
(a) peanut oil;
(b) fully hydrogenated peanut oil; and
(c) modified peanut oil.
(5) [Repealed, SOR/2011-28, s. 2]
- SOR/78-728, s. 1
- SOR/79-23, s. 6
- SOR/79-662, s. 1
- SOR/88-559, s. 4
- SOR/92-626, s. 8
- SOR/93-145, s. 2
- SOR/93-465, s. 1
- SOR/95-548, s. 5(F)
- SOR/97-263, s. 1
- SOR/2000-417, s. 1
- SOR/2010-143, s. 39(E)
- SOR/2011-28, s. 2
- SOR/2022-143, s. 8
- SOR/2022-168, s. 4
- SOR/2022-169, s. 6
B.01.010 (1) In this section, common name includes a name set out in the Common Names for Ingredients and Components Document.
(2) An ingredient or component shall be shown in the list of ingredients by its common name.
(3) For the purposes of subsection (2),
(a) the ingredient or component set out in the Common Names for Ingredients and Components Document shall be shown in the list of ingredients by the common name of that ingredient or component that is set out in that Document; and
(b) the ingredient or component set out in the Common Names for Ingredients and Components Document may be shown in the list of ingredients by the common name of that ingredient or component that is set out in that Document.
(4) Despite subsection (2) and subsection B.01.008.2(5), if a food contains ingredients of the same class, those ingredients may be shown by a class name if
(a) they consist of more than one component and are not listed in the table to subsection B.01.009(1); and
(b) their components are shown
(i) immediately after the class name of the ingredients of which they are components, in such a manner as to indicate that they are components of the ingredients, and
(ii) in descending order of their collective proportion by weight of those ingredients.
- SOR/79-23, ss. 7, 8(F)
- SOR/79-529, s. 3
- SOR/80-632, s. 1
- SOR/84-300, ss. 4(E), 5(F)
- SOR/91-124, s. 2
- SOR/92-626, s. 9
- SOR/92-725, s. 1
- SOR/93-243, s. 2(F)
- SOR/93-465, s. 2
- SOR/95-548, s. 5(F)
- SOR/97-516, s. 1
- SOR/98-458, ss. 1, 7(F)
- SOR/2005-98, s. 7
- SOR/2007-302, s. 4(F)
- SOR/2011-28, s. 3
- SOR/2016-305, s. 6
- SOR/2021-46, s. 3(E)
- SOR/2022-143, s. 9
B.01.010.1 (1) The following definitions apply in this section and in sections B.01.010.2 to B.01.010.4.
- food allergen
food allergen means any protein from any of the following foods, or any modified protein, including any protein fraction, that is derived from any of the following foods:
(a) almonds, Brazil nuts, cashews, hazelnuts, macadamia nuts, pecans, pine nuts, pistachios or walnuts;
(b) peanuts;
(c) sesame seeds;
(d) wheat or triticale;
(e) eggs;
(f) milk;
(g) soybeans;
(h) crustaceans;
(i) molluscs;
(j) fish; or
(k) mustard seeds. (allergène alimentaire)
- food allergen source, gluten source and added sulphites statement
food allergen source, gluten source and added sulphites statement means a statement appearing on the label of a prepackaged product that indicates the source of a food allergen or gluten that is present in the product or the presence in the product of added sulphites in a total amount of 10 p.p.m. or more. (mention des sources d’allergènes alimentaires ou de gluten et des sulfites ajoutés)
- gluten
gluten means
(a) any gluten protein from the grain of any of the following cereals or from the grain of a hybridized strain that is created from at least one of the following cereals:
(i) barley,
(ii) oats,
(iii) rye,
(iv) triticale,
(v) wheat; or
(b) any modified gluten protein, including any gluten protein fraction, that is derived from the grain of any of the cereals referred to in paragraph (a) or from the grain of a hybridized strain referred to in that paragraph. (gluten)
- height
height, in respect of type, means the height of the lower case letter “x”. (hauteur)
(2) If a food allergen or gluten is present in a prepackaged product, the source of the food allergen or gluten, as the case may be, must be shown on the label of the product in
(a) the list of ingredients; or
(b) in a food allergen source, gluten source and added sulphites statement.
(3) Subsection (2) does not apply to a food allergen or gluten that is present in a prepackaged product as a result of cross-contamination.
(4) Subsection (2) does not apply to a to a food allergen or gluten that is present in a prepackaged product referred to in paragraphs B.01.008(2)(a) to (e) unless a list of ingredients is shown on the product’s label.
(5) [Repealed, SOR/2019-98, s. 1]
(6) The source of a food allergen required to be shown under subsection (2) must be shown
(a) for a food allergen from a food referred to in one of paragraphs (a), (b) and (e) of the definition food allergen in subsection (1) or derived from that food, by the name of the food as shown in the applicable paragraph, expressed in the singular or plural;
(b) for a food allergen from the food referred to in paragraph (c) of the definition food allergen in subsection (1) or derived from that food, by the name “sesame”, “sesame seed” or “sesame seeds”;
(c) for a food allergen from a food referred to in one of paragraphs (d) and (f) of the definition food allergen in subsection (1) or derived from that food, by the name of the food as shown in the applicable paragraph;
(d) for a food allergen from the food referred to in paragraph (g) of the definition food allergen in subsection (1) or derived from that food, by the name “soy”, “soya”, “soybean” or “soybeans”;
(e) for a food allergen from a food referred to in one of paragraphs (h) to (j) of the definition food allergen in subsection (1) or derived from that food, by the common name of the food that is set out in the Common Names for Ingredients and Components Document; and
(f) for a food allergen from the food referred to in paragraph (k) of the definition food allergen in subsection (1) or derived from that food, by the name “mustard”, “mustard seed” or “mustard seeds”.
(7) The source of gluten required to be shown under subsection (2) must be shown
(a) for gluten from the grain of a cereal referred to in one of subparagraphs (a)(i) to (v) of the definition gluten in subsection (1) or derived from that grain, by the name of the cereal as shown in the applicable subparagraph; and
(b) for gluten from the grain of a hybridized strain created from one or more of the cereals referred to in subparagraphs (a)(i) to (v) of the definition gluten in subsection (1) or derived from that grain, by the names of the cereals as shown in the applicable subparagraphs.
(8) For the purpose of paragraph (2)(a), the source of the food allergen or gluten must be shown in the list of ingredients, in parentheses, as follows:
(a) immediately after the ingredient that is shown in that list, if the food allergen or gluten
(i) is that ingredient,
(ii) is present in that ingredient, but is not a component of or present in a component of that ingredient, or
(iii) is, or is present in, a component of that ingredient and the component is not shown in the list of ingredients; or
(b) immediately after the component that is shown in the list of ingredients, if the food allergen or gluten is that component or is present in that component.
(9) Despite subsection (2), the source of the food allergen or gluten must be shown on the label of the product in the food allergen source, gluten source and added sulphites statement if the food allergen or gluten
(a) is, or is present in, an ingredient that is not shown in the list of ingredients, but is not a component of that ingredient or present in a component of that ingredient; or
(b) is, or is present in, a component and neither the component nor the ingredient in which it is present is shown in the list of ingredients.
(10) Despite subsection (8), the source of the food allergen or gluten is not required to be shown in parentheses immediately after the ingredient or component, as the case may be, if the source of the food allergen or gluten appears
(a) in the list of ingredients
(i) as part of the common name of the ingredient or component, or
(ii) in parentheses, under subsection (8), immediately after another ingredient or component; or
(b) in the food allergen source, gluten source and added sulphites statement.
(11) For greater certainty, nothing in subsection (8) affects how an ingredient or component may be shown in the list of ingredients under paragraph B.01.010(3)(b).
- SOR/2011-28, s. 4
- SOR/2016-305, ss. 7, 72
- SOR/2019-98, s. 1
- SOR/2021-46, s. 4(E)
- SOR/2022-143, s. 10
- SOR/2022-168, s. 5
B.01.010.2 (1) In this section and in section B.01.010.3, sulphites means the food additives that are set out in the Common Names for Ingredients and Components Document and are present in a prepackaged product.
(2) For greater certainty, the definition sulphites in subsection (1) includes only sulphites that are present in the prepackaged product as a result of being added.
(3) If sulphites are present in a prepackaged product in a total amount of 10 parts per million or more and none are required to be shown in the list of ingredients under section B.01.008 or B.01.009, the sulphites must be shown on the label of the product in
(a) the list of ingredients; or
(b) the food allergen source, gluten source and added sulphites statement that complies with the requirements of subsection B.01.010.3(1).
(4) Subsection (3) does not apply to sulphites present in the prepackaged products referred to in paragraphs B.01.008(2)(a) to (e) unless a list of ingredients is shown on the product’s label.
(5) [Repealed, SOR/2019-98, s. 2]
(6) Sulphites that are shown on a label of the product under subsection (3) must be shown as follows:
(a) if the sulphites are shown in the list of ingredients,
(i) by one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”, or
(ii) individually by the name of the food additive that is set out in the Common Names for Ingredients and Components Document, except that the name “sodium dithionite”, “sulphur dioxide” or “sulphurous acid” must be followed, in parentheses, by one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”; or
(b) if the sulphites are shown in a food allergen source, gluten source and added sulphites statement, by one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”.
(7) Sulphites that are shown in the list of ingredients under paragraph (6)(a) must be shown as follows:
(a) sulphites that are a component of an ingredient that is shown in the list of ingredients must be shown either in parentheses immediately after the ingredient or at the end of that list where they may be shown in any order with the other ingredients that are shown at the end of that list in accordance with subsection B.01.008.2(4); and
(b) in all other cases, the sulphites must be shown at the end of the list of ingredients where they may be shown in any order with the other ingredients that are shown at the end of that list in accordance with subsection B.01.008.2(4).
(8) If sulphites are present in a prepackaged product in a total amount of 10 parts per million or more and any of them are required to be shown in the list of ingredients under section B.01.008 or B.01.009, in the case of sulphites shown individually by the name “sodium dithionite”, “sulphur dioxide” or “sulphurous acid”, that name must be followed, in parentheses, by one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”.
(9) If the total amount of sulphites present in the prepackaged product is 10 parts per million or more, sulphites that are required to be shown in a list of ingredients under section B.01.008 or B.01.009 may also be shown on the label of the product in a food allergen source, gluten source and added sulphites statement that complies with the requirements of subsection B.01.010.3(1).
(10) Despite subparagraph (6)(a)(ii) and subsection (8), if sulphites are shown individually in a list of ingredients, by the name “sodium dithionite”, “sulphur dioxide” or “sulphurous acid”, that name is not required to be followed, in parentheses, by one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents” if
(a) in the list of ingredients,
(i) the term “sulfite” or “sulphite” appears in the common name of another sulphite, or
(ii) one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents” is shown in parentheses following another sulphite; or
(b) one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents” is shown in a food allergen source, gluten source and added sulphites statement on the label of the product.
- SOR/2011-28, s. 4
- SOR/2016-305, ss. 8, 72
- SOR/2019-98, s. 2
- SOR/2022-143, s. 11
- SOR/2022-168, s. 6
B.01.010.3 (1) A food allergen source, gluten source and added sulphites statement must
a) be introduced by a title that
(i) consists of the terms
(A) “Contains”, “Contains:” or “Contains :” in the English version of the statement, and
(B) “Contient”, “Contient :” or “Contient :” in the French version of the statement,
(ii) is shown in bold type,
(iii) is shown without any intervening printed, written or graphic material appearing between it and the rest of the statement, and
(iv) if the statement is preceded by a statement referred to in subsection B.01.014(1) and begins on the line on which that statement ends, is shown in a type that is of a height that is at least 0.2 mm greater than the height of the type used in that statement;
(a.1) appear, in respect of each linguistic version, immediately after any statement referred to in subsection B.01.014(1) appearing in the same language or, if there is no such statement, immediately after the list of ingredients appearing in the same language and, in either case, without any intervening printed, written or graphic material;
(a.2) appear on the same continuous surface as the statement or list that immediately precedes it and be shown in the same manner as the list of ingredients is shown under subsection B.01.008.2(2);
(b) include all of the following information, even if all or part of that information is also shown in the list of ingredients:
(i) the source for each food allergen that is present in the prepackaged product,
(ii) each source for the gluten that is present in the product, and
(iii) one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”, if the total amount of sulphites present in the product is 10 parts per million or more; and
(c) show information entries in regular or bold type, using lower case letters except for the first letter of each entry, which shall be an upper case letter, and shall use a bullet point or comma to separate each entry.
(2) Despite paragraph (1)(b), the following information is not required to be shown in the statement more than once:
(a) the same source of a food allergen;
(b) the same source of gluten; and
(c) one of the common names “sulfites”, “sulfiting agents”, “sulphites” or “sulphiting agents”.
(3) If the English and French versions of the statement appear on the same continuous surface of the label, the version that follows the other version shall not begin on the same line as that on which the other version ends, except in the case of a prepackaged product that has an available display surface of less than 100 cm2.
- SOR/2011-28, s. 4
- SOR/2016-305, s. 9
- SOR/2022-168, s. 7
B.01.010.4 (1) If the label of a prepackaged product includes a declaration alerting consumers that, due to a risk of cross-contamination, the product may contain the source of a food allergen or gluten,
(a) the declaration must appear immediately after
(i) the food allergen source, gluten source and added sulphites statement, if there is one,
(ii) any statement referred to in subsection B.01.014(1), if no food allergen source, gluten source and added sulphites statement appears on the label, or
(iii) the list of ingredients, if neither of the statements referred to in subparagraph (ii) appears on the label;
(b) the declaration must appear in both English and French if the statement or list immediately preceding it appears in both languages;
(c) the declaration must appear on the same continuous surface as the statement or list that immediately precedes it and be shown in the same manner as the list of ingredients is shown under subsection B.01.008.2(2);
(d) the declaration must appear without any intervening printed, written or graphic material between it and the statement or list that immediately precedes it, except that a solid line may appear before the declaration if the declaration begins on a different line than the line on which the statement or list ends;
(e) the declaration must be shown in bold type if it begins on the line on which the statement or list that immediately precedes it ends and if it is not introduced by a title;
(f) any title that introduces the declaration must be shown in bold type if the declaration begins on the line on which the statement or list that immediately precedes it ends; and
(g) if the declaration is preceded by a statement referred to in subsection B.01.014(1) and begins on the line on which the statement ends, the title of the declaration — or the declaration itself, if no title appears — must be shown in a type that is of a height that is at least 0.2 mm greater than the height of the type used in the statement.
(2) If the English and French versions of the declaration appear on the same continuous surface of the label, the version that follows the other version must not begin on the line on which the other version ends unless the prepackaged product has an available display surface of less than 100 cm2.
- SOR/2016-305, s. 10
- SOR/2022-168, s. 8
B.01.011 (1) Where it is an acceptable manufacturing practice for a manufacturer to
(a) omit from his prepackaged product any food that is ordinarily an ingredient or component, or
(b) substitute in whole or in part in his prepackaged product any other food for a food that is ordinarily an ingredient or component,
the list of ingredients for the 12-month period commencing from the time the label is applied to the prepackaged product may show as ingredients or components the foods that may be omitted and the foods that may be used as substitutes if
(c) all the foods that may be used as ingredients or components throughout the 12-month period are shown in the list of ingredients;
(d) it is clearly stated as part of the list of ingredients that the food shown as an ingredient or component may not be present or that another food may be substituted for a food shown as an ingredient or component; and
(e) the foods that may be omitted or substituted are grouped with the same class of foods that are used as ingredients or components and the foods within each such group are listed in descending order of the proportion by weight in which they will probably be used during the 12-month period.
(2) Where it is an acceptable manufacturing practice for a manufacturer to vary the proportions of ingredients or components of his prepackaged product, the list of ingredients for the 12-month period commencing from the time the label is applied to the prepackaged product may show the ingredients or components in the same proportions throughout the 12-month period if
(a) it is clearly stated as part of the list of ingredients that the proportions indicated are subject to change; and
(b) the ingredients or components are listed in descending order of the proportion by weight in which they will probably be used during the 12-month period.
B.01.012 (1) In this section,
- local government unit
local government unit means a city, metropolitan government area, town, village, municipality or other area of local government but does not include any local government unit situated within a bilingual district established under the Official Languages Act; (collectivité locale)
- local food
local food means a food that is manufactured, processed, produced or packaged in a local government unit and sold only in
(a) the local government unit in which it is manufactured, processed or packaged,
(b) one or more local government units that are immediately adjacent to the one in which it is manufactured, processed, produced or packaged, or
(c) the local government unit in which it is manufactured, processed, produced or packaged and in one or more local government units that are immediately adjacent to the one in which it is manufactured, processed, produced or packaged; (produit alimentaire local)
- mother tongue
mother tongue means the language first learned in childhood by persons in any area of Canada and still understood by them as ascertained by the decennial census taken immediately preceding the date on which the food referred to in subsection (3) is sold to the consumer; (langue maternelle)
- official languages
official languages means the English language and the French language; (langues officielles)
- specialty food
specialty food means a food — other than a human milk fortifier or supplemented food — that
(a) has special religious significance and is used in religious ceremonies; or
(b) is an imported food
(i) that is not widely used by the population as a whole in Canada, and
(ii) for which there is no readily available substitute that is manufactured, processed, produced or packaged in Canada and that is generally accepted as being a comparable substitute; (aliment spécial)
- test market food
test market food means a food that, prior to the date of the notice of intention respecting that food referred to in subsection (5), was not sold in Canada in that form and that differs substantially from any other food sold in Canada with respect to its composition, function, state or packaging form and includes a food referred to in section B.01.054. (produit alimentaire d’essai)
(2) Subject to subsections (9), (10) and (11), all information required by these Regulations to be shown on the label of a food shall be shown in both official languages.
(3) Subject to subsections (4) to (6), subsection (2) does not apply to a local food or test market food if
(a) it is sold in a local government unit in which one of the official languages is the mother tongue of less than 10 per cent of the total number of persons residing in the local government unit; and
(b) the information required by these Regulations to be shown on the label of a food is shown in the official language that is the mother tongue of at least 10 per cent of the total number of persons residing in the local government unit.
(4) Where one of the official languages is the mother tongue of less than 10 per cent of the total number of persons residing in a local government unit and the other official language is the mother tongue of less than 10 per cent of the total number of persons residing in the same local government unit, subsection (3) does not apply.
(5) Subsection (3) does not apply to a test market food unless the person who intends to conduct the test marketing of the food has, six weeks prior to conducting the test marketing, filed with the President of the Canadian Food Inspection Agency a notice of intention in a form acceptable to the President.
(6) A test market food shall, for the purposes of subsection (3), cease to be a test market food upon the expiration of 12 cumulative months after the date on which it was first offered for sale as a test market food but any test market food that was acquired for resale by a person, other than the person who filed the notice of intention referred to in subsection (5), before the expiration of those 12 cumulative months, shall continue to be a test market food for the purposes of subsection (3) until it is sold.
(7) Subsection (2) does not apply to a specialty food if the information required by these Regulations to be shown on the label thereon is shown in one of the official languages.
(8) Where there are one or more surfaces on the label of a food that are of at least the same size and prominence as the principal display panel, the information required by these Regulations to be shown on the principal display panel may be shown in one official language if such information is shown in the other official language on one of those other surfaces.
(9) Subsection (2) does not apply to the identity and principal place of business of the person by or for whom the food was manufactured, processed, produced or packaged for resale if this information is shown in one of the official languages.
(10) Subsection (2) does not apply to the following common names if the common name appears on the principal display panel in the following manner:
- Scotch Whisky
- Irish Whisky
- Highland Whisky
- Dry Gin
- Bourbon
- Tennessee Whisky
- Tequila
- Mezcal
- Rye Whisky
- Crème de Menthe
- Crème de Cacao
- Crème de Cassis
- Crème de Banane
- Triple Sec
- Anisette
- Crème de Noyau
- Brandy
- Sake or Saki
- Advocaat or Advokaat
- Kirsch
- Slivovitz
- Ouzo
- Cherry Brandy Liqueur
- Kummel
- Akvavit
- Aquavit
- Armagnac
- Marc
- Grappa
- Calvados
- Poire William
- Crème de Bleuets
- Curaçao Orange
- Liqueur de Fraise
- Mandarinette
- Prunelle de Bourgogne
- Chartreuse
- Pastis
- Fior d’Alpe
- Strega
- Campari
- Americano
- Apricot Brandy Liqueur
- Peach Brandy Liqueur
- Sloe Gin
- Manhattan
- Martini
(11) Subsection (2) does not apply to the label of a shipping container destined to a commercial or industrial enterprise or an institution, if
(a) the shipping container and its contents are not resold as a one unit prepackaged product to a consumer at the retail level; and
(b) all information required by these Regulations to be shown on a label of a food is shown in one of the official languages.
- SOR/79-23, s. 9
- SOR/79-529, s. 4
- SOR/84-300, s. 6
- SOR/93-603, s. 1
- SOR/95-548, s. 5
- SOR/2000-184, s. 62
- SOR/2016-305, s. 11
- SOR/2021-57, s. 6
- SOR/2022-169, s. 7
B.01.013 (1) Unless specifically required by the Act or these Regulations, no reference, direct or indirect, to the Act or to these Regulations shall be made on any label of, or in any advertisement for, a food.
(2) Notwithstanding subsection (1), where a food complies with a standard established by these Regulations and the manufacturer of the food has substantiated, by means of the results of tests carried out before the statement is made or by other evidence that exists before the statement is made, that the food so complies, a statement that the food “complies with the standard for (naming the common name of the food in respect of which the claim is made) in the Food and Drug Regulations” may be made on the label of, or in an advertisement for, the food.
- SOR/92-626, s. 10
- SOR/95-548, s. 5(F)
B.01.014 (1) The label of a food that contains aspartame must carry a statement warning individuals with phenylketonuria that the food contains phenylalanine or a statement to the effect that aspartame contains phenylalanine.
(2) The statement must
(a) be shown in bold type;
(b) appear, in respect of each linguistic version, immediately after the list of ingredients appearing in the same language, either on the same line as the last ingredient in the list or on the following line, without any intervening printed, written or graphic material; and
(c) appear on the same continuous surface as the list of ingredients and be shown in the same manner as that list is shown under subsection B.01.008.2(2).
- SOR/81-617, s. 2
- SOR/88-559, s. 5
- SOR/2003-11, s. 6
- SOR/2016-305, s. 75(F)
- SOR/2018-108, s. 400
- SOR/2022-168, s. 9
B.01.015 [Repealed, SOR/2022-168, s. 9]
B.01.016 [Repealed, SOR/2022-168, s. 9]
B.01.017 [Repealed, SOR/2022-168, s. 9]
B.01.018 The label of a food that contains polydextrose shall indicate the amount of polydextrose expressed in grams per serving of stated size.
- SOR/93-276, s. 2
- SOR/94-779, s. 1
- SOR/97-512, s. 1
- SOR/2003-11, s. 10
- SOR/2016-305, s. 75(F)
B.01.019 [Repealed, SOR/2022-168, s. 10]
B.01.020 [Repealed, SOR/2022-168, s. 10]
B.01.021 (1) The label of a food that contains erythritol shall carry a statement indicating the amount of erythritol expressed in grams per serving of stated size unless the label carries a nutrition facts table or supplemented food facts table.
(2) The statement of the amount of erythritol shall be grouped together with the statement of the amount of any other sugar alcohols and the amount of polydextrose.
- SOR/2004-261, s. 1
- SOR/2016-305, s. 75(F)
- SOR/2022-169, s. 8
B.01.022 [Repealed, SOR/2022-168, s. 11]
B.01.023 The label of a food that is a table-top sweetener that contains aspartame, sucralose, acesulfame-potassium or neotame must carry a statement of the sweetness per serving expressed in terms of the amount of sugar required to produce an equivalent degree of sweetness.
- SOR/2007-176, s. 4
- SOR/2016-305, s. 75(F)
- SOR/2018-108, s. 400
- SOR/2022-168, s. 11
B.01.033 (1) Except in the case of a formulated liquid diet, human milk fortifier or human milk substitute, it is prohibited to sell a food represented in any manner as containing hydrolyzed or partially hydrolyzed collagen, hydrolyzed or partially hydrolyzed gelatin or hydrolyzed or partially hydrolyzed casein unless the label carries the following statement on the principal display panel in the same size type used for the common name:
“CAUTION, DO NOT USE AS SOLE SOURCE OF NUTRITION”.
(2) In this section, formulated liquid diet means a food that meets the requirements of sections B.24.101 to B.24.103.
- SOR/78-65, s. 1
- SOR/2021-57, s. 7
B.01.034 [Repealed, SOR/88-559, s. 7]
B.01.035 (1) Subject to subsection (8), where an irradiated food referred to in column 1 of the table to Division 26 is offered for sale as a prepackaged product, the principal display panel of the label applied to the package shall carry the symbol described in subsection (5).
(2) Where an irradiated food referred to in column 1 of the table to Division 26 is not a prepackaged product and is offered for sale, a sign that carries the symbol described in subsection (5) shall be displayed immediately next to the food.
(3) The symbol required pursuant to subsection (1) or (2) shall appear in close proximity on the principal display panel referred to in subsection (1) or on the sign referred to in subsection (2) to one of the following statements or a written statement that has the same meaning:
(a) “treated with radiation”;
(b) “treated by irradiation”; or
(c) “irradiated”.
(4) No person shall sell a food referred to in column 1 of the table to Division 26 that has been irradiated in the manner set out in subsection B.26.003(2) unless the requirements of subsections (1) to (3) are met.
(5) For the purposes of subsections (1) to (3), the symbol that indicates the irradiated food shall
(a) have an outer diameter
(i) in the case referred to in subsection (1), equal to or greater than the height of the numerical quantity prescribed by paragraph 229(1)(a) and subsections 229(2) and (3) of the Safe Food for Canadians Regulations for the declaration of net quantity of the package, and
(ii) in the case referred to in subsection (2), not less than 5 cm; and
(b) be in the following form:

(6) Notwithstanding subsection B.01.009(1), any food referred to in column 1 of the table to Division 26 that is an ingredient or component of a prepackaged product and that has been irradiated shall, if the food constitutes 10 per cent or more of the prepackaged product, be included in the list of ingredients and preceded by the statement “irradiated”.
(7) The label attached to a shipping container that contains a food set out in column 1 of the table to Division 26 that has been subjected to the maximum absorbed dose set out in column 5 shall carry the statement that is required by subsection (3) and the statement “Do not irradiate again.”.
(8) Where a shipping container constitutes the package of the prepackaged product, the label attached to the shipping container shall carry the statement required by subsection (7) but need not carry the symbol required by subsection (5).
(9) Any advertising of an irradiated food referred to in column 1 of the table to Division 26 shall identify the food as having been irradiated.
(10) The statements referred to in subsections (3) and (6) to (8) shall be in both official languages in accordance with subsection B.01.012(2).
- SOR/89-172, s. 1
- SOR/2017-16, s. 1
- SOR/2018-108, s. 400
B.01.037 [Repealed, SOR/88-559, s. 8]
B.01.040 [Repealed, SOR/88-559, s. 9]
B.01.042 Where a standard for a food is prescribed in this Part,
(a) the food shall contain only the ingredients included in the standard for the food;
(b) each ingredient shall be incorporated in the food in a quantity within any limits prescribed for that ingredient; and
(c) if the standard includes an ingredient to be used as a food additive for a specified purpose, that ingredient shall be a food additive set out in one of the tables to section B.16.100 for use as an additive to that food for that purpose.
B.01.043 Subject to section B.25.062, where a standard for a food is not prescribed in this Part,
(a) the food shall not contain any food additives except food additives set out in a table to section B.16.100 for use as additives to that food for the purpose set out in that table; and
(b) each such food additive shall be incorporated in the food in a quantity within any limits prescribed for that food and food additive in that table.
- SOR/87-640, s. 1
B.01.044 Where the limit prescribed for a food additive in a table to section B.16.100 is stated to be “Good Manufacturing Practice”, the amount of the food additive added to a food in manufacturing and processing shall not exceed the amount required to accomplish the purpose for which that additive is permitted to be added to that food.
B.01.045 A food additive shall,
(a) where specifications are set out in this Part for that additive, meet those specifications;
(b) where no specifications are set out in this Part for the additive, meet the specifications for it, if any, set out in
(i) the Food Chemicals Codex, tenth edition, 2016, published by the United States Pharmacopeial Convention, Rockville, MD, United States of America, as that or any subsequent edition, including their supplements, may be amended from time to time; or
(ii) the Combined Compendium of Food Additive Specifications, prepared by the Joint FAO/WHO Expert Committee on Food Additives and published by the Food and Agriculture Organization of the United Nations, on the website of the Organization, as amended from time to time; and
(c) in the case of a food colour for which no specifications exist under paragraph (a) or (b), contain no more than
(i) 3 parts per million of arsenic, and
(ii) 10 parts per million of lead.
(d) [Repealed, SOR/2010-142, s. 1]
(e) [Repealed, SOR/97-512, s. 2]
(f) [Repealed, SOR/2016-305, s. 12]
(g) [Repealed, SOR/97-512, s. 2]
- SOR/82-383, s. 1
- SOR/91-527, s. 3
- SOR/92-93, s. 1
- SOR/92-551, s. 1
- SOR/93-276, s. 3
- SOR/94-625, s. 4
- SOR/94-779, s. 2
- SOR/95-172, s. 2
- SOR/97-512, s. 2
- SOR/2010-142, s. 1
- SOR/2016-305, s. 12
B.01.046 [Repealed, SOR/2016-74, s. 2]
B.01.047 [Repealed, SOR/2016-74, s. 2]
B.01.047.1 (1) The following definitions apply in this section.
- BSE
BSE means Bovine Spongiform Encephalopathy. (ESB)
- specified risk material
specified risk material means
(a) the skull, brain, trigeminal ganglia, eyes, tonsils, spinal cord and dorsal root ganglia of cattle aged 30 months or older; and
(b) the distal ileum of cattle of all ages. (matériel à risque spécifié)
(2) No person shall sell or import for sale food that contains specified risk material.
(3) Subsection (2) does not apply in respect of food that originates from a country that is designated as being free from BSE in accordance with section 7 of the Health of Animals Regulations.
(4) Subsection (2) does not apply in respect of food that is packaged for sale or imported for sale before the day on which this subsection comes into force.
- SOR/2003-265, s. 1
B.01.048 (1) No person shall sell
(a) any animal intended for consumption as food if any product containing any drug listed in subsection (2) has been administered to the animal;
(b) any food that is derived from an animal if any product containing a drug listed in subsection (2) has been administered to the animal; or
(c) any food that is derived from an animal if the food contains any residue of a drug listed in subsection (2).
(2) The drugs referred to in subsection (1) are
(a) chloramphenicol and its salts and derivatives;
(b) a 5-nitrofuran compound;
(c) clenbuterol and its salts and derivatives;
(d) a 5-nitroimidazole compound; and
(e) diethylstilbestrol and other stilbene compounds.
- SOR/85-685, s. 1
- SOR/87-626, s. 1
- SOR/94-568, s. 1
- SOR/97-510, s. 1
- SOR/2003-292, s. 1
- SOR/2016-74, s. 3
B.01.049 No person shall use, in labelling, packaging, advertising or selling a food that does not meet the requirements of the kashruth applicable to it, the word “kosher” or any letters of the Hebrew alphabet or any other word, expression, depiction, sign, symbol, mark, device or other representation that indicates or that is likely to create an impression that the food is kosher.
- SOR/84-300, s. 8
B.01.050 A person must not use, in labelling, packaging, advertising or selling a food, the word “halal” — or any letters of the Arabic alphabet or any other word, expression, depiction, sign, symbol, mark, device or other representation that indicates or that is likely to create an impression that the food is halal — unless the name of the person or body that certified the food as halal is indicated on the label or package or in the advertisement or sale.
- SOR/2014-76, s. 1
B.01.053 No person shall sell a product represented as a ready breakfast or instant breakfast or by any similar designation unless each serving of stated size of the product contains
(a) not less than 4.0 mg. iron;
(b) Vitamin A, thiamine, riboflavin, niacin or niacinamide and Vitamin C;
(c) a good dietary source of protein; and
(d) where consumed as directed, not less than 300 calories.
- SOR/2003-11, s. 13
- SOR/2016-305, s. 75(F)
B.01.054 (1) In order to generate information in support of an amendment to the Regulations, the Minister may issue to the manufacturer or distributor of a food, where the food or the packaging, labelling or advertising of the food does not comply with the requirements of these Regulations, a Temporary Marketing Authorization Letter that authorizes the sale of the food described therein or the packaging, labelling or advertising of the food described therein for a specified period of time, within a designated area and in a specified quantity, in the manner specified in the Letter if
(a) the manufacturer or distributor of the food has supplied to the Minister the following information:
(i) the purpose for which the temporary marketing authorization of the food is required,
(ii) a description of the food including a sample and proposed label,
(iii) a description of any proposed variation from the requirements of these Regulations,
(iv) adequate data to show that the use of the food will not be detrimental to the health of the purchaser or user,
(v) the proposed quantity of the food to be sold,
(vi) the proposed period of time required for such sale,
(vii) the proposed area designated for such sale, and
(viii) such other data as the Minister may require; and
(b) the manufacturer or distributor of the food has agreed to
(i) describe the food on a label or in an advertisement in a manner that is not false, misleading or deceptive,
(ii) use such marks or statements on the label or in any advertisement as the Minister may require,
(iii) on request, submit to the Minister results of the temporary marketing, and
(iv) on request, withdraw the product from sale where the Minister is of the opinion that it is in the public interest to do so.
(2) The Minister shall, in any Temporary Marketing Authorization Letter issued pursuant to subsection (1), set out
(a) the common name and description of the food to be sold;
(b) the name and address of the manufacturer or distributor of the food;
(c) the purpose for which the temporary marketing of the food is authorized;
(d) the quantity of the food that is authorized for sale;
(d.1) the type of packaging, labelling or advertising authorized in respect of the food where the Letter is intended to authorize a variation from a requirement of any provision of the Regulations respecting packaging, labelling or advertising;
(e) the period of time during which the food may be sold; and
(f) the designated area within which the food may be sold.
- SOR/81-566, s. 1
- SOR/85-275, s. 1
- SOR/2018-69, s. 27
B.01.055 (1) A manufacturer or distributor named in a Temporary Marketing Authorization Letter issued pursuant to subsection B.01.054(1) may, for the purpose set out in the Letter, sell the food in the manner authorized in the Letter and package, label or advertise that food in the manner authorized in the Letter for the period of time, within the designated area and in the quantity set out in the Letter.
(2) No provision of these Regulations made pursuant to paragraph 30(1)(b) of the Act applies in respect of a food or the packaging, labelling or advertising of a food for which a Temporary Marketing Authorization Letter has been issued pursuant to subsection B.01.054(1) to the extent that the food, or the packaging, labelling or advertising of the food, as authorized in the Letter, does not comply with that provision.
- SOR/81-566, s. 1
- SOR/85-275, s. 2
- SOR/90-814, s. 4
B.01.056 [Repealed, SOR/2016-74, s. 4]
B.01.060 to B.01.066 [Repealed, SOR/88-559, s. 10]
B.01.070 [S]. Mixed nuts or a mixture of nuts shall consist of a mixture of nuts in which not less than five per cent by weight of each type of nuts is present in the mixture.
B.01.071 Where a prepackaged product is a mixture of nuts, the percentage and common name of the nut that is present in the product in the greatest amount by weight shall be applied to the principal display panel of the package in close proximity to the common name of the product.
- SOR/88-336, s. 3
- SOR/92-626, s. 11
B.01.072 Notwithstanding any requirement prescribed in Part B, a food product that has been subjected to heat in the presence of a vaporized liquid solution of smoke derived from hardwood, hardwood sawdust or corn cobs may be described as “smoked”.
- SOR/92-626, s. 11
B.01.080 (1) In this section, frozen means preserved by freezing temperature and does not include any surface freezing that may occur during holding and transportation.
(2) Where meat, meat by-products, poultry meat, poultry by-products or fish, or meat of any marine or fresh water animal, that has been frozen is thawed prior to sale, the words “previously frozen” shall be shown
(a) on the principal display panel in close proximity to the common name of the food and in letters at least as legible and conspicuous as those used in the common name;
(b) anywhere on the principal display panel in letters of not less than 1/4 of an inch (6.4 millimetres) in height; or
(c) on a sign displayed adjacent to the food in letters that are easily visible and legible to a prospective purchaser.
(3) Where part of a food referred to in subsection (2) has been frozen and thawed prior to sale, the words “Made from fresh and frozen portions” or “Made from fresh and frozen (naming the food)” shall be shown in the manner described in paragraph (2)(a), (b) or (c).
- SOR/88-336, s. 3
- SOR/2022-143, s. 13(E)
B.01.090 (1) No person shall offer for sale at retail any solid cut meat or solid cut poultry meat to which phosphate salts or water has been added, unless that meat or poultry meat is contained in a package and carries a label.
(2) The label referred to in subsection (1) shall contain a statement of the minimum percentage of meat protein as part of the common name of the product on the principal display panel of the package in type that is as legible and conspicuous as any other type on that display panel, and in letters that are at least one half of the size of the letters used in the common name of the product but that are not less than 1.6 mm in height.
- SOR/94-262, s. 1
B.01.091 The label of any solid cut meat or solid cut poultry meat that has had phosphate salts or water added to it, that is not cured and that is prepackaged at retail shall contain a statement of the ingredients contained in the food in accordance with subsections B.01.008.2(1) to (5) and (7).
- SOR/94-262, s. 1
- SOR/2003-11, s. 14
- SOR/2016-305, s. 13
B.01.092 Sections B.01.090 and B.01.091 do not apply in respect of side bacon, Wiltshire bacon, pork jowls, salt pork or salt beef.
- SOR/94-262, s. 1
B.01.100 (1) The common name of a simulated meat product or simulated poultry product shall be the common name of the meat product or poultry product that is simulated, modified by the word “simulated”.
(2) The word “simulated” in the common name of a simulated meat product or simulated poultry product shall be shown in letters of at least the same size and prominence as those used in the remainder of the common name of that product.
(3) Where a simulated meat product or a simulated poultry product is not a prepackaged product, the common name of the product and the other information required by this section to be shown on the label of a simulated meat product or simulated poultry product shall be shown on a sign displayed on or adjacent to the product in letters that are legible and conspicuous to a prospective purchaser.
(4) The words
(a) “contains no meat”, in the case of a simulated meat product, and
(b) “contains no poultry”, in the case of a simulated poultry product,
shall be shown on the principal display panel of the label of a simulated meat product or simulated poultry product in close proximity to the common name and in letters of at least the same size and prominence as those shown in the common name.
(5) to (7) [Repealed, SOR/88-559, s. 11]
- SOR/88-336, s. 3
- SOR/88-559, s. 11
B.01.101 (1) For the purposes of this section and section B.01.102, source of protein means any food that contains protein, but does not include spices, seasonings, flavours, artificial flavours, flavour enhancers, food additives and similar foods that contain only small amounts of protein.
(2) The common name of a meat product extender shall be the common name of each food in the meat product extender that is a source of protein, plus
(a) the word “meat”, or the common name of the meat product that is to be extended, plus the word “extender”; or
(b) the words “extender for” plus the common name of the meat product that is to be extended.
(3) The common name of a poultry product extender shall be the common name of each food in the poultry product extender that is a source of protein, plus
(a) the word “poultry”, or the common name of the poultry product that is to be extended plus the word “extender”; or
(b) the words “extender for” plus the common name of the poultry product that is to be extended.
(4) Foods that are a source of protein in the meat product extender or poultry product extender shall be shown by their common names in the common name of that meat product extender or poultry product extender
(a) in descending order of their proportion by weight of the meat product extender or poultry product extender; and
(b) in letters of at least the same size and prominence as those used in the remainder of the common name of the meat product extender or poultry product extender.
(5) and (6) [Repealed, SOR/88-559, s. 12]
- SOR/88-559, s. 12
- SOR/2022-143, s. 14
B.01.102 (1) The common name of an extended meat product or an extended poultry product shall be the common name of the meat product or poultry product that is extended, modified by the common name of each of the foods that are sources of protein in the extended meat product or extended poultry product.
(2) Notwithstanding subsection (1),
(a) the word or words “meat”, “meat product”, “poultry”, “poultry meat” or “poultry meat by-product” as the case may be, may be used in the common name of an extended meat product or extended poultry product as the common name of the food therein that is a source of protein derived from a meat product or poultry product; and
(b) where it is an acceptable manufacturing practice for a manufacturer to omit from his meat product extender or poultry product extender any source of protein derived from a plant that is ordinarily an ingredient of that meat product extender or poultry product extender, or to substitute in whole or in part in his meat product extender or poultry product extender any source of protein derived from a plant for a source of protein that is ordinarily an ingredient of that meat product extender or poultry product extender, the word “plant” may be used in the common name of an extended meat product or extended poultry product as the common name of the food therein that is a source of protein derived from a plant.
(3) Foods that are a source of protein in an extended meat product or extended poultry product shall be shown by their common names in the common name of that product
(a) in descending order of their proportion by weight of that product; and
(b) in letters of at least the same size and prominence as those used in the remainder of the common name of that product.
(4) Where an extended meat product or extended poultry product is not a prepackaged product, the common name of that product and the information required by this section to be shown on the label of an extended meat product or extended poultry product shall be shown on a sign displayed on or adjacent to that product in letters that are legible and conspicuous to a prospective purchaser.
(5) to (7) [Repealed, SOR/88-559, s. 13]
- SOR/84-300, s. 9
- SOR/88-559, s. 13
- SOR/2022-143, s. 15
B.01.103 (1) [Repealed, SOR/2022-143, s. 16]
(2) to (4) [Repealed, SOR/88-559, s. 14]
- SOR/88-559, s. 14
- SOR/2022-143, s. 16
B.01.300 [Repealed, SOR/2003-11, s. 15]
B.01.301 (1) No person shall, on the label of or in any advertisement for a food, other than in the nutrition facts table or supplemented food facts table, if any, include a declaration of the food’s energy value or the amount of a nutrient or supplemental ingredient contained in the food unless it is declared in the following manner, per serving of stated size:
(a) in the case of the energy value, in Calories;
(b) in the case of a vitamin referred to in subsection D.01.002(1) that is not one of the following vitamins, in the applicable unit set out in subsection D.01.003(1):
(i) beta-carotene as a form of vitamin A or retinol, including its derivatives, as a form of vitamin A, or both, if either is a supplemental ingredient, and
(ii) niacin, if it is a supplemental ingredient;
(b.01) in the case of the following vitamins, in the applicable unit set out in column 3 of the List of Permitted Supplemental Ingredients:
(i) beta-carotene as a form of vitamin A or retinol, including its derivatives, as a form of vitamin A, or both, if either is a supplemental ingredient, and
(ii) niacin, if it is a supplemental ingredient;
(b.1) in the case of the following mineral nutrients:
(i) for sodium, potassium, calcium, phosphorus, magnesium, iron, zinc, chloride, copper and manganese, in milligrams, and
(ii) for iodide, chromium, selenium and molybdenum, in micrograms;
(c) in the case of cholesterol, in milligrams;
(d) in the case of the mineral ion content of prepackaged water or ice, in parts per million;
(d.1) in the case of a supplemental ingredient other than a vitamin or mineral nutrient referred to in this subsection, in the applicable unit referred to in column 3 of the List of Permitted Supplemental Ingredients; and
(e) in any other case, in grams.
(1.1) Despite subsection D.01.003(1) and for the purposes of this section, the amount in metric units of the vitamins referred to in paragraph (1)(b.01) must be determined in terms of their amount in the supplemented food in accordance with column 5 of the List of Permitted Supplemental Ingredients, as applicable.
(2) Despite subsection (1), a person may, on the label of or in any advertisement for a food, other than in the nutrition facts table or supplemented food facts table, if any, include a declaration of the percentage of the daily value of a nutrient contained in the food if
(a) in the case of a food that is not a supplemented food, the nutrient is listed in column 1 of the table to section B.01.401 or the table to section B.01.402 and the percentage of the daily value of the nutrient is required or permitted to be declared in the nutrition facts table;
(b) in the case of a supplemented food, the nutrient is listed in column 1 of the table to section B.29.002 or the table to section B.29.003, or referred to in column 2 of item 18 of the table to section B.29.002, and the percentage of the daily value of the nutrient is required or permitted to be declared in the supplemented food facts table; and
(c) the percentage of the daily value of the nutrient is declared per serving of stated size.
(3) A declaration referred to in subsection (1) or (2) that appears on the label of a food shall be
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the food may be shown in that language only and is shown on the label in that language.
- SOR/88-559, s. 15
- SOR/2003-11, s. 16
- SOR/2016-305, ss. 14, 75(F)
- SOR/2022-169, s. 9
B.01.302 If the label of a multiple-serving prepackaged product indicates that the product contains or, if prepared as directed in or on the package, provides a specified number of servings or portions, that information must be based on the serving of stated size set out in the nutrition facts table or supplemented food facts table, as the case may be.
- SOR/2016-305, s. 15
- SOR/2018-108, s. 394
- SOR/2022-169, s. 10
B.01.303 and B.01.304 [Repealed, SOR/2003-11, s. 17]
B.01.305 (1) No person shall, on the label of or in any advertisement for a food, make a representation, express or implied, respecting a protein unless the food meets the conditions set out in column 2 of item 8 of the Table of Permitted Nutrient Content Statements and Claims for the subject “source of protein” set out in column 1.
(2) No person shall, on the label of or in any advertisement for a food, other than a supplemented food, make a representation, express or implied, respecting an amino acid unless
(a) the food meets the conditions set out in column 2 of item 8 of the Table of Permitted Nutrient Content Statements and Claims for the subject “source of protein” set out in column 1; and
(b) the label or advertisement includes a declaration of the amount of histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan and valine contained in the food, expressed in grams per serving of stated size.
(3) Subsections (1) and (2) do not apply in respect of
(a) a formulated liquid diet, human milk fortifier, human milk substitute or food represented as containing a human milk substitute;
(b) foods represented for use in gluten-free diets, protein restricted diets, low (naming the amino acid) diets and (naming the amino acid) free diets;
(c) the word “protein” when used as part of the common name of an ingredient in the list of ingredients;
(d) the declaration of amino acids in the list of ingredients;
(e) the common names that are set out in the Common Names for Ingredients and Components Document, if shown in the list of ingredients in accordance with paragraph B.01.010(3)(a);
(f) the common name of a single amino acid preparation that may be sold as a food;
(g) any statement referred to in subsection B.01.014(1);
(h) a statement or claim set out in column 4 of item 7 of the Table of Permitted Nutrient Content Statements and Claims respecting the subject “low in protein” set out in column 1;
(i) a declaration of the amount of protein in the nutrition facts table or supplemented food facts table, as the case may be;
(j) a statement of the protein content of a food as required by paragraph B.24.103(c), subparagraph B.24.202(a)(ii), paragraph B.24.304(b) or B.25.057(1)(a) or subparagraph B.25.057(2)(c)(i) or (d)(i); or
(k) a statement that a food is not a source of protein.
(4) A representation referred to in subsection (1) or (2) that appears on the label of a food shall be
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the food may be shown in that language only and is shown on the label in that language.
- SOR/88-559, s. 15
- SOR/90-830, s. 3(F)
- SOR/2003-11, s. 18
- SOR/2016-305, s. 75(F)
- SOR/2021-57, s. 8
- SOR/2022-143, s. 17
- SOR/2022-168, s. 12
- SOR/2022-168, s. 52
- SOR/2022-169, s. 11
B.01.306 to B.01.310 [Repealed, SOR/2003-11, s. 19]
B.01.311 (1) Subject to subsections (2) and (3), no person shall, on the label of or in any advertisement for a food, make a representation, express or implied, concerning the action or effect of the food’s energy value or of a nutrient contained in the food.
(2) The label of or advertisement for a food may carry a statement or claim set out in column 1 of the table following section B.01.603.
(3) Subject to section B.01.312, the label of or advertisement for a food may carry a statement or claim to the effect that the food’s energy value or a nutrient contained in the food is generally recognized as an aid in maintaining the functions of the body necessary to the maintenance of good health and normal growth and development.
(4) If a statement or claim described in subsection (3) concerns a nutrient not listed in column 1 of the tables to sections B.01.401 and B.01.402 — or, as the case may be, not listed in column 1 of the tables to sections B.29.002 and B.29.003 and not referred to in column 2 of item 18 of the table to section B.29.002 — the amount of the nutrient contained in the food must be expressed on any part of the label in grams per serving of stated size.
(5) A statement or claim referred to in subsection (2) or (3) that appears on the label of a food shall be
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the food may be shown in that language only and is shown on the label in that language.
- SOR/88-559, s. 15
- SOR/2003-11, s. 20
- SOR/2016-305, s. 75(F)
- SOR/2022-169, s. 12
B.01.312 (1) If a statement or claim described in subsection B.01.311(3) is made on the label of or in an advertisement for a food that is not a prepackaged product or in an advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the label or advertisement shall include a declaration, per serving of stated size, of
(a) the energy value, if the energy value is the subject of the statement or claim; or
(b) the amount of the nutrient, if a nutrient is the subject of the statement or claim.
(1.1) Subsection (1) does not apply if the statement or claim is made on the label of or in the advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
(2) If the statement or claim is made in an advertisement other than a radio or television advertisement, the declaration referred to in subsection (1) shall be
(a) adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(b) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
(3) If the statement or claim is made in a radio advertisement or in the audio portion of a television advertisement, the declaration referred to in subsection (1) shall immediately precede or follow the statement or claim.
(4) If the statement or claim is made in a television advertisement, the declaration referred to in subsection (1) shall be communicated
(a) in the audio mode, if the statement or claim is made only in the audio portion of the advertisement or in both the audio and visual portions; or
(b) in the audio or visual mode, if the statement or claim is made only in the visual portion of the advertisement.
(5) If the declaration referred to in subsection (1) is communicated in the visual mode of a television advertisement, it shall
(a) appear concurrently with and for at least the same amount of time as the statement or claim;
(b) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(c) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 16, 75(F)
Nutrition Symbols
Mandatory Information
B.01.350 (1) Except as otherwise provided in this section, the principal display panel of a prepackaged product must carry a symbol that is set out in Schedule K.1 if
(a) the product, as offered for sale, contains a nutrient that is set out in column 1 of the table to this section; and
(b) the amount of the nutrient, calculated as a percentage of the daily value, meets or exceeds the applicable threshold set out in columns 2 to 7 of that table.
(2) For the purposes of subsection (1), the percentage of the daily value for the nutrient is calculated on the basis of the amount of the nutrient, by weight, per serving of stated size or per reference amount, whichever is greater.
(3) Despite subsection (2), if the prepackaged product requires reconstitution with water or another liquid or the addition of any other ingredient for its preparation and the reference amount applicable to the product only refers to the food in its prepared form, the percentage of the daily value for the nutrient is calculated on the basis of the amount of the nutrient, by weight, per serving of stated size.
(4) In the case of a prepackaged product referred to in subsection (3), the applicable threshold set out in columns 2 to 7 of the table to this section is determined on the basis of the product’s serving of stated size rather than its reference amount.
(5) Subsection (1) does not apply to the following:
(a) shipping containers, unless the containers and their contents are sold as a one unit prepackaged product to a consumer at the retail level;
(b) prepackaged products with an available display surface of less than 15 cm2;
(c) prepackaged individual portions of food that are intended solely to be served by a restaurant or other commercial enterprise with meals or snacks;
(d) ready-to-serve multiple-serving prepackaged products that are intended solely to be served in a commercial or industrial enterprise or an institution;
(e) prepackaged products that are intended solely for use as an ingredient in the manufacture of other prepackaged products intended for sale to a consumer at the retail level or as an ingredient in the preparation of food by a commercial or industrial enterprise or an institution;
(f) milk, partly skimmed milk, skim milk, goat’s milk, partly skimmed goat’s milk, skimmed goat’s milk, (naming the flavour) milk, (naming the flavour) partly skimmed milk, (naming the flavour) skim milk or cream sold in a refillable glass container;
(g) sweetening agents, including maple sugar and maple syrup;
(h) salt for table or general household use, celery salt, garlic salt, onion salt and any other seasoning salt if “salt” forms part of the common name of the food;
(i) fats and oils referred to in Division 9, fish and other marine fats and oils, butter, ghee and margarine and other similar substitutes for butter; or
(j) individual rations intended for use by military personnel engaged in operations or exercises.
(6) Subsection (1) does not apply, in respect of a nutrient, to the following prepackaged products if the only ingredients containing the nutrient are, in the case of saturated fat or sodium, ingredients set out in subsection (7) or, in the case of sugars, ingredients set out in subsection (8):
(a) whole or cut fruits or vegetables, including frozen, canned or dried fruits or vegetables;
(b) milk obtained from any animal, in liquid or powdered form;
(c) whole eggs, including liquid, frozen or dried eggs, or whole egg mixes;
(d) nuts, seeds or nut or seed butters in which less than 30% of the total fat content consists of saturated fat;
(e) vegetable oils or marine oils in which less than 30% of the total fat content consists of saturated fat;
(f) marine and fresh water animal products referred to in Division 21 in which less than 30% of the total fat content consists of saturated fat; or
(g) any combination of the foods referred to in paragraphs (a) to (f).
(7) For the purposes of subsection (6) in relation to saturated fat and sodium, the ingredients are the following ingredients to which no saturated fat or sodium has been added:
(a) whole or cut fruits or vegetables, including frozen, canned or dried fruits or vegetables;
(b) milk obtained from any animal, in liquid or powdered form;
(c) whole eggs, including liquid, frozen or dried eggs, or whole egg mixes;
(d) nuts, seeds or nut or seed butters in which less than 30% of the total fat content consists of saturated fat;
(e) vegetable oils or marine oils in which less than 30% of the total fat content consists of saturated fat; and
(f) marine and fresh water animal products referred to in Division 21 in which less than 30% of the total fat content consists of saturated fat.
(8) For the purposes of subsection (6) in relation to sugars, the ingredients are the following ingredients to which no sugars have been added:
(a) the ingredients set out in paragraphs (7)(a), (b) and (d);
(b) dairy products other than those referred to in paragraph (7)(b);
(c) grains; and
(d) legumes.
(9) Subsection (1) does not apply, in respect of saturated fat and sugars, to prepackaged products that are cheese or yogurt – including drinkable yogurt – that are made from dairy products, kefir or buttermilk unless
(a) in the case of saturated fat, the product contains an ingredient, other than any of the following ingredients, that contains saturated fat:
(i) milk ingredients,
(ii) modified milk ingredients,
(iii) nuts or seeds in which less than 30% of the total fat content consists of saturated fat,
(iv) vegetable oils or marine oils in which less than 30% of the total fat content consists of saturated fat, or
(v) marine and fresh water animal products referred to in Division 21 in which less than 30% of the total fat content consists of saturated fat; and
(b) in the case of sugars, the product contains an ingredient, other than any of the following ingredients, that contains sugars or the product contains any of the following ingredients to which sugars have been added:
(i) whole or cut fruits or vegetables, including frozen, canned or dried fruits or vegetables,
(ii) dairy products,
(iii) grains,
(iv) legumes, or
(v) nuts or seeds.
(10) For the purposes of paragraphs (6)(a) and (7)(a), fruit does not include coconut.
(11) Subsection (1) does not apply, in respect of sodium, to prepackaged products that are cheese made from dairy products.
(12) For the application of subsections (9) and (11), the prepackaged products must
(a) have a reference amount of 30 g or 30 mL or less and contain 10% or more of the daily value for calcium per serving of stated size or per reference amount, whichever is greater; or
(b) have a reference amount greater than 30 g or 30 mL and contain 15% or more of the daily value for calcium per serving of stated size or per reference amount, whichever is greater.
(13) Subsection (1) does not apply to the following prepackaged products unless they are required to carry a nutrition facts table on their label:
(a) a beverage with an alcohol content of more than 0.5%;
(b) a raw single ingredient meat, meat by-product, poultry meat or poultry meat by-product that is not ground;
(c) a raw single ingredient marine or fresh water animal product;
(d) a product sold only in the retail establishment where it is prepared and processed from its ingredients, including from a pre-mix if an ingredient other than water is added to the pre-mix during the preparation and processing of the product;
(e) a product sold only at a road-side stand, craft show, flea market, fair, farmers’ market or sugar bush by the individual who prepared and processed the product;
(f) an individual serving that is sold for immediate consumption and that has not been subjected to a process to extend its durable life, including special packaging;
(g) a product sold only in the retail establishment where it is packaged, if it is labelled by means of a sticker and has an available display surface of less than 200 cm2; or
(h) a product with an available display surface of less than 100 cm2.
(13.01) Subsection (1) does not apply to a prepackaged product that is a raw single ingredient meat, meat by-product, poultry meat or poultry meat by-product that is ground unless it meets any of the conditions that are set out in paragraph B.01.401(3)(a), (b) or (e).
(13.1) Subject to subsection (13.2), subsection (1) does not apply to a supplemented food that has an available display surface of less than 100 cm2 other than a supplemented food that is referred to in paragraph B.29.018(2)(b).
(13.2) Subsection (1) does not apply to a supplemented food that has an available display surface of less than 100 cm2, whether or not the supplemented food is referred to in paragraph B.29.018(2)(b), if the supplemented food carries a supplemented food caution identifier.
(14) If, as a result of the application of any provision in subsections (5) to (13.2), subsection (1) does not apply to a prepackaged product or does not apply to a prepackaged product in respect of a particular nutrient, that provision prevails over any other provision in subsections (5) to (13.2) that indicates otherwise.
(15) Despite any other provision in this section, the label of the following prepackaged products must not carry a symbol referred to in subsection (1):
(a) a prepackaged product intended solely for infants six months of age or older but less than one year of age;
(b) a human milk fortifier;
(c) a human milk substitute or a food represented as containing a human milk substitute;
(d) a formulated liquid diet as defined in section B.24.001;
(e) a meal replacement;
(f) a nutritional supplement;
(g) a food represented for protein-restricted diets;
(h) a food represented for low (naming the amino acid) diets; and
(i) a food represented for use in a very low energy diet as defined in section B.24.001.
TABLE
Thresholds Requiring a Nutrition Symbol
Column 1 Column 2 Column 3 Column 4 Column 5 Column 6 Column 7 Nutrient Threshold for a prepackaged product other than one referred to in columns 5 to 7 Threshold for a prepackaged product intended solely for children one year of age or older, but less than four years of age Item Prepackaged product with a reference amount greater than 30 g or 30 mL, unless the product is described in column 4 Prepackaged product with a reference amount of 30 g or 30 mL or less Prepackaged main dish with a reference amount of 200 g or more Prepackaged product with a reference amount greater than 30 g or 30 mL, unless the product is described in column 7 Prepackaged product with a reference amount of 30 g or 30 mL or less Prepackaged main dish with a reference amount of 170 g or more 1 Saturated fat 15% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 3 of Part 1 of the Table of Daily Values 10% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 3 of Part 1 of the Table of Daily Values 30% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 3 of Part 1 of the Table of Daily Values 15% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 2 of Part 1 of the Table of Daily Values 10% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 2 of Part 1 of the Table of Daily Values 30% of the daily value for the sum of saturated fatty acids and trans fatty acids set out in column 2 of Part 1 of the Table of Daily Values 2 Sugars 15% of the daily value for sugars set out in column 3 of Part 1 of the Table of Daily Values 10% of the daily value for sugars set out in column 3 of Part 1 of the Table of Daily Values 30% of the daily value for sugars set out in column 3 of Part 1 of the Table of Daily Values 15% of the daily value for sugars set out in column 2 of Part 1 of the Table of Daily Values 10% of the daily value for sugars set out in column 2 of Part 1 of the Table of Daily Values 30% of the daily value for sugars set out in column 2 of Part 1 of the Table of Daily Values 3 Sodium 15% of the daily value for sodium set out in column 3 of Part 1 of the Table of Daily Values 10% of the daily value for sodium set out in column 3 of Part 1 of the Table of Daily Values 30% of the daily value for sodium set out in column 3 of Part 1 of the Table of Daily Values 15% of the daily value for sodium set out in column 2 of Part 1 of the Table of Daily Values 10% of the daily value for sodium set out in column 2 of Part 1 of the Table of Daily Values 30% of the daily value for sodium set out in column 2 of Part 1 of the Table of Daily Values
Presentation of Nutrition Symbol
B.01.351 (1) A nutrition symbol must be displayed in black and white and must be in accordance with the applicable symbol set out in Schedule K.1.
(2) Subject to subsection (3), a nutrition symbol must be presented in one of the following formats:
(a) unilingual horizontal format, where two separate versions of the symbol are shown, one in English (EH) and one in French (FH); or
(b) bilingual horizontal format (BH), where the symbol is shown in both official languages.
(3) If the principal display surface is less than or equal to 450 cm2 and the width of the nutrition symbol in either of the horizontal formats that could apply exceeds the width of the principal display panel, the symbol must be presented in one of the following formats:
(a) unilingual vertical format, where two separate versions of the symbol are shown, one in English (EV) and one in French (FV); or
(b) bilingual vertical format (BV), where the symbol is shown in both official languages.
(4) If, in accordance with subsection B.01.012(3) or (7), the information required by these Regulations may be shown on the label of a prepackaged product in English only or in French only and is shown in that language, the nutrition symbol may be displayed on the principal display panel of the product in that language only on a continuous surface of the available display surface.
(5) If a nutrition symbol is presented in a bilingual format, the order in which the languages appear may be reversed from the order shown in the applicable symbol set out in Schedule K.1.
B.01.352 (1) The version of the nutrition symbol that must be carried on a prepackaged product that has a principal display surface that is within a range set out in column 1 of the table to this section and that contains a nutrient set out in column 2 in an amount that meets or exceeds the applicable threshold referred to in subsection B.01.350(1) is the version that is referred to in column 3 or 5 of the table or, if applicable, column 4 or 6 of the table.
(2) Despite subsection (1), a prepackaged product that has a principal display surface that is greater than 250 cm2 may carry the applicable version of the nutrition symbol that is referred to in item 4, column 2, of Table 1 or 3 in the Directory of Nutrition Symbol Specifications if the product is sold only in the retail establishment where it is packaged and is labelled by means of a sticker.
(3) A nutrition symbol must be displayed in accordance with the applicable specifications set out in the Directory of Nutrition Symbol Specifications.
(4) Despite subsection (3), a nutrition symbol may be displayed with larger dimensions than those set out in column 3 of the applicable table in the Directory of Nutrition Symbol Specifications if the symbol is enlarged in a proportional manner vertically and horizontally.
TABLE
Nutrition Symbols and Formats
Column 1 Column 2 Column 3 Column 4 Column 5 Column 6 Item Range of principal display surface Nutrients that meet or exceed threshold in subsection B.01.350(1) Nutrition symbol in unilingual horizontal format Nutrition symbol in unilingual vertical format Nutrition symbol in bilingual horizontal format Nutrition symbol in bilingual vertical format 1 > 30 cm2 Saturated fat (Sat fat), sugars and sodium 1(EH) and 1(FH) 1(EV) and 1(FV) 1(BH) 1(BV) Saturated fat (Sat fat) and sugars 2(EH) and 2(FH) 2(EV) and 2(FV) 2(BH) 2(BV) Sugars and sodium 3(EH) and 3(FH) 3(EV) and 3(FV) 3(BH) 3(BV) Saturated fat (Sat fat) and sodium 4(EH) and 4(FH) 4(EV) and 4(FV) 4(BH) 4(BV) Saturated fat (Sat fat) 5(EH) and 5(FH) 5(EV) and 5(FV) 5(BH) 5(BV) Sugars 6(EH) and 6(FH) 6(EV) and 6(FV) 6(BH) 6(BV) Sodium 7(EH) and 7(FH) 7(EV) and 7(FV) 7(BH) 7(BV) 2 ≤ 30 cm2 Saturated fat (Sat fat), sugars and sodium 1(EH) and 1(FH) 1(EV) and 1(FV) 1(BH) 1(BV) Saturated fat (Sat fat) and sugars 8(EH) and 8(FH) 8(EV) and 8(FV) 8(BH) 8(BV) Sugars and sodium 9(EH) and 9(FH) 9(EV) and 9(FV) 9(BH) 9(BV) Saturated fat (Sat fat) and sodium 10(EH) and 10(FH) 10(EV) and 10(FV) 10(BH) 10(BV) Saturated fat (Sat fat) 11(EH) and 11(FH) 11(EV) and 11(FV) 11(BH) 11(BV) Sugars 12(EH) and 12(FH) 12(EV) and 12(FV) 12(BH) 12(BV) Sodium 13(EH) and 13(FH) 13(EV) and 13(FV) 13(BH) 13(BV)
B.01.353 (1) Subject to subsection (2), if a prepackaged product contains an assortment of foods and one or more of the foods requires a nutrition symbol, the nutrition symbols must be displayed in a manner that clearly indicates the applicable nutrients that are contained in each food.
(2) If a prepackaged product contains ingredients that are intended to be combined together or foods that are intended to be consumed together, the nutrition symbol must display the nutrients that are contained in the product as a whole.
B.01.354 The characters and other elements of a nutrition symbol must not touch each other.
Location of Nutrition Symbol
B.01.355 (1) A nutrition symbol must be displayed
(a) in the case of a prepackaged product with a principal display panel whose height is less than its width, on the right half of the panel; and
(b) in the case of any other prepackaged product, on the upper half of the principal display panel.
(2) The nutrition symbol must be surrounded by a buffer that
(a) has a width that is equal to or greater than that set out in column 4 of the applicable table in the Directory of Nutrition Symbol Specifications; and
(b) does not contain any text or other graphic material.
(3) If a prepackaged product is cylindrical in shape, the outer edge of the buffer must be a minimum distance of 10% of the width of the principal display surface from the edge of the left or right side of that surface.
(4) If it is impossible to comply with both paragraph (1)(a) and subsection (3), the nutrition symbol may be displayed partially in the left half of the principal display panel but only to the extent necessary to comply with that subsection.
B.01.356 A nutrition symbol must be oriented in the same manner as most of the other information that appears on the principal display panel unless the panel is displayed in the vertical plane and most of the other information is not displayed parallel with the base of the package, in which case the symbol must be oriented in such a manner that the words appearing in it are parallel with the base.
Prominence of Health-related Representations
B.01.357 (1) If both a nutrition symbol and a health-related representation appear on the principal display panel of a prepackaged product and the representation relates to a nutrient that is referred to in the nutrition symbol,
(a) the height of the upper case letters in the representation must not exceed the height of the upper case letters, excluding any accents, in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”; and
(b) the height of the tallest ascender of the lower case letters in the representation must not exceed the height of the tallest ascender of the lower case letters in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”.
(2) If both a nutrition symbol and a health-related representation appear on the principal display panel of a prepackaged product and the representation does not relate to a nutrient that is referred to in the nutrition symbol,
(a) the height of the upper case letters in the representation must not exceed two times the height of the upper case letters, excluding any accents, in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”; and
(b) the height of the tallest ascender of the lower case letters in the representation must not exceed two times the height of the tallest ascender of the lower case letters in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”.
(3) In this section, health-related representation means
(a) a declaration referred to in subsection B.01.301(1) or (2);
(b) a statement or claim referred to in subsection B.01.311(2) or (3);
(c) a representation referred to in any of sections B.01.503 to B.01.513;
(d) a statement or claim referred to in subsection B.01.601(1); or
(e) any other health-related statement, logo, symbol, seal of approval or mark, other than
(i) the brand name or product name of a prepackaged product, or
(ii) a statement or claim referred to in any of sections D.01.004 to D.01.007 and D.02.002 to D.02.005.
Prohibitions — Resemblance to Nutrition Symbol
B.01.358 (1) It is prohibited to
(a) label a prepackaged product with any representation, including a word, phrase, illustration, sign, mark, symbol or design, that is likely to be mistaken for a nutrition symbol; or
(b) advertise or sell a prepackaged product that is labelled with a representation referred to in paragraph (a).
(2) For the purposes of subsection (1), a representation does not include a supplemented food caution identifier.
Interpretation
B.01.400 (1) For the purpose of sections B.01.401 to B.01.603, fat means all fatty acids expressed as triglycerides.
(2) For the purpose of sections B.01.401 to B.01.603, the amount of vitamins shall be determined in accordance with section D.01.003.
- SOR/2003-11, s. 20
- SOR/2016-305, s. 17
Nutrition Labelling
Core Information
B.01.401 (1) Except as otherwise provided in this section and sections B.01.402 to B.01.406 and B.01.467 to B.01.469, the label of a prepackaged product shall carry a nutrition facts table that contains only the information set out in column 1 of the table to this section, expressed using a description set out in column 2, in the unit set out in column 3 and in the manner set out in column 4.
(1.1) For the purpose of subsection (1), the serving of stated size set out in a nutrition facts table for a prepackaged product, as expressed in a metric unit, shall be used as the basis for determining the information appearing in the nutrition facts table in respect of the energy value and nutrient content of the product.
(1.2) The percentage of the daily value for a mineral nutrient shown in the nutrition facts table for a prepackaged product in accordance with subsection (1) shall be established on the basis of the amount, by weight, of the mineral nutrient per serving of stated size for the product, rounded off in the applicable manner set out in column 4 of the table to this section.
(2) Subsection (1) does not apply to a prepackaged product if
(a) all the information set out in column 1 of the table to this section, other than in respect of item 1 (“Serving of stated size”), may be expressed as “0” in the nutrition facts table in accordance with this section;
(b) the product is
(i) a beverage with an alcohol content of more than 0.5%,
(ii) [Repealed, SOR/2016-305, s. 18]
(iii) a raw single ingredient meat, meat by-product, poultry meat or poultry meat by-product,
(iv) a raw single ingredient marine or fresh water animal product,
(v) sold only in the retail establishment where the product is prepared and processed from its ingredients, including from a pre-mix if an ingredient other than water is added to the pre-mix during the preparation and processing of the product,
(vi) sold only at a road-side stand, craft show, flea market, fair, farmers’ market or sugar bush by the individual who prepared and processed the product,
(vii) an individual serving that is sold for immediate consumption and that has not been subjected to a process to extend its durable life, including special packaging, or
(viii) sold only in the retail establishment where the product is packaged, if the product is labelled by means of a sticker and has an available display surface of less than 200 cm2; or
(c) the product is
(i) a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating,
(ii) a prepackaged individual portion of food that is intended solely to be served by a restaurant or other commercial enterprise with meals or snacks, or
(iii) milk, partly skimmed milk, skim milk, goat’s milk, partly skimmed goat’s milk, skimmed goat’s milk, (naming the flavour) milk, (naming the flavour) partly skimmed milk, (naming the flavour) skim milk or cream sold in a refillable glass container.
(3) Despite paragraphs (2)(a) and (b), subsection (1) applies to a prepackaged product if
(a) the product contains an added vitamin or mineral nutrient;
(b) a vitamin or mineral nutrient is declared as a component of one of the product’s ingredients other than flour;
(c) [Repealed, SOR/2022-168, s. 14]
(d) the product is a meat, meat by-product, poultry meat or poultry meat by-product that is ground; or
(e) the label of the product, or any advertisement for the product that is made or placed by or on the direction of the manufacturer of the product, contains
(i) a reference to the energy value, a nutrient set out in column 1 of the table to this section or in column 1 of the table to section B.01.402 or a constituent of such a nutrient, other than information required by Division 12 or a reference to the common name of an ingredient in the list of ingredients for the product,
(ii) a representation that expressly or implicitly indicates that the product has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004,
(iii) a health-related name, statement, logo, symbol, seal of approval or mark, or
(iv) the phrase “nutrition facts”, “valeur nutritive” or “valeurs nutritives”.
(4) Subsection (1) does not apply to a formulated liquid diet, human milk fortifier, human milk substitute, food represented as containing a human milk substitute, meal replacement, nutritional supplement, food represented for use in a very low energy diet or supplemented food.
(5) The label of, or an advertisement for, a formulated liquid diet, human milk fortifier, human milk substitute, food represented as containing a human milk substitute, meal replacement, nutritional supplement, food represented for use in a very low energy diet or supplemented food shall not contain a nutrition facts table or the phrase “nutrition facts”, “valeur nutritive” or “valeurs nutritives”.
(6) If, for a prepackaged product other than one intended solely for infants six months of age or older but less than one year of age, the information in respect of six or more of the energy value and nutrients referred to in column 1 of items 2 to 5 and 7 to 15 of the table to this section may be expressed as “0” in the nutrition facts table in accordance with this section, the nutrition facts table need only include the following information:
(a) the serving of stated size;
(b) the energy value;
(c) the amount of fat;
(d) the amount of carbohydrate;
(e) the amount of protein;
(f) the amount of any nutrient that is the subject of a representation referred to in subparagraph (3)(e)(ii);
(g) the amount of any sugar alcohol, vitamin or mineral nutrient added to the prepackaged product, other than iodide added to salt for table or general household use or fluoride added to prepackaged water or ice;
(h) the amount of any vitamin or mineral nutrient that is declared as a component of one of the prepackaged product’s ingredients other than flour;
(i) the amount of any nutrient referred to in column 1 of any of items 4, 5, 7, 8, 10, 11 and 13 to 15 of the table to this section that may not be expressed as “0” in the nutrition facts table; and
(j) the statement “Not a significant source of (naming each nutrient that is omitted from the nutrition facts table in accordance with this subsection)” or, if the prepackaged product meets the condition specified in subsection B.01.455(3), the statement “Not a significant source of other nutrients”.
(6.1) The nutrition facts table of a single-serving prepackaged product, other than one that is a prepackaged meal, need only include the following information:
(a) the serving of stated size;
(b) the energy value;
(c) the amount of fat;
(d) the amount of carbohydrate;
(e) the amount of protein;
(f) the amount of any nutrient that is the subject of a representation referred to in subparagraph (3)(e)(ii);
(g) the amount of any sugar alcohol, vitamin or mineral nutrient added to the prepackaged product, other than iodide added to salt for table or general household use or fluoride added to prepackaged water or ice;
(h) the amount of any nutrient referred to in column 1 of item 4 or 5 of the table to this section and the sum of saturated fatty acids and trans fatty acids if any of the amounts or the sum may not be expressed as “0” in the nutrition facts table; and
(i) the amount of any nutrient referred to in column 1 of item 8 or 11 of the table to this section that may not be expressed as “0” in the nutrition facts table;
(j) the % Daily Value interpretative statement.
(7) Subsection (1) does not apply to a prepackaged product
(a) that is intended solely for use as an ingredient in the manufacture of other prepackaged products intended for sale to a consumer at the retail level or as an ingredient in the preparation of food by a commercial or industrial enterprise or an institution; or
(b) that is a multiple-serving ready-to-serve prepackaged product intended solely to be served in a commercial or industrial enterprise or an institution.
(8) If the nutrition facts table on the label of a prepackaged product corresponds to Figure 6.5(B), 6.6(B), 6.5.1(B), 6.6.1(B), 7.3(B), 7.4(B), 7.3.1(B), 7.4.1(B), 17.2(E) and (F), 17.2.1(E) and (F), 24.1 (E) and (F) to 24.6(E) and (F), 25.1 (B) to 25.6(B), 26.1(B) to 26.4(B), 32.1(E) and (F) or 32.2(E) and (F) of the Directory of NFT Formats, the nutrition facts table is not required to show the % Daily Value interpretative statement referred to in item 16 of the table to this section.
TABLE
Core Information
Item Column 1 Column 2 Column 3 Column 4 Information Description Unit Manner of expression 1 Serving of stated size “Serving Size (naming the serving size)”, “Serving (naming the serving size)” or “Per (naming the serving size)” The size is expressed
(a) in the case of a single-serving prepackaged product,
(i) per package, and
(ii) in grams or millilitres, in accordance with subparagraph B.01.002A(2)(a)(i) or (ii); and
(b) in the case of a multiple-serving prepackaged product, in the following units set out in column 3B of the Table of Reference Amounts:
(i) the household measure that applies to the product, and
(ii) the metric measure that applies to the product.
(1) The size when expressed in a metric unit is rounded off
(a) if it is less than 10 g or 10 mL, to the nearest multiple of 0.1 g or 0.1 mL; and
(b) if it is 10 g or more or 10 mL or more, to the nearest multiple of 1 g or 1 mL.
(2) The size when expressed as a fraction is represented by a numerator and a denominator separated by a line.
(3) The size shall include the word “assorted” if the information in the nutrition facts table of a prepackaged product that contains an assortment of foods is set out as a composite value.
2 Energy value “Calories”, “Total Calories” or “Calories, Total” The value is expressed in Calories per serving of stated size. The value is rounded off
(a) if it is less than 5 Calories
(i) if the product meets the conditions set out in column 2 of item 1 of the Table of Permitted Nutrient Content Statements and Claims for the subject” free of energy” set out in column 1, to “0” Calorie, and
(ii) in all other cases, to the nearest multiple of 1 Calorie;
(b) if it is 5 Calories or more but not more than 50 Calories, to the nearest multiple of 5 Calories; and
(c) if it is more than 50 Calories, to the nearest multiple of 10 Calories.
3 Amount of fat “Fat”, “Total Fat” or “Fat, Total” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g
(i) if the product meets the conditions set out in column 2 of item 11 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of fat” set out in column 1 and the amounts of saturated fatty acids and trans fatty acids are declared as “0 g” in the nutrition facts table or are omitted from that table in accordance with subsection B.01.401(6) and no other fatty acids are declared in an amount greater than 0 g, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to “0%“; and
(b) in all other cases, to the nearest multiple of 1%.
4 Amount of saturated fatty acids “Saturated Fat”, “Saturated Fatty Acids”, “Saturated” or “Saturates” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g
(i) if the product meets the conditions set out in column 2 of item 18 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of saturated fatty acids” set out in column 1, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
5 Amount of trans fatty acids “Trans Fat”, “Trans Fatty Acids” or “Trans” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g
(i) if the product meets the conditions set out in column 2 of item 22 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of trans fatty acids” set out in column 1, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
6 The sum of saturated fatty acids and trans fatty acids “Saturated Fat + Trans Fat”, “Saturated Fatty Acids + Trans Fatty Acids”, “Saturated + Trans” or “Saturates + Trans” The sum is expressed as a percentage of the daily value per serving of stated size. The percentage is rounded off
(a) if the amounts of saturated fatty acids and trans fatty acids are declared as “0 g”, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
7 Amount of cholesterol “Cholesterol” The amount
(a) is expressed in milligrams per serving of stated size; and
(b) may also be expressed as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if the product meets the conditions set out in column 2 of item 27 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of cholesterol” set out in column 1, to “0 mg”; and
(b) in all other cases, to the nearest multiple of 5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg” to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
8 Amount of sodium “Sodium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg
(i) if the product meets the conditions set out in column 2 of item 31 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of sodium or salt” set out in column 1, to “0 mg”, and
(ii) in all other cases, to the nearest multiple of 1 mg;
(b) if it is 5 mg or more but not more than 140 mg, to the nearest multiple of 5 mg; and
(c) if it is more than 140 mg, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
9 Amount of carbohydrate “Carbohydrate”, “Total Carbohydrate” or “Carbohydrate, Total” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
10 Amount of fibre “Fibre”, “Fiber”, “Dietary Fibre” or “Dietary Fiber” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
11 Amount of sugars “Sugars” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
12 Amount of protein “Protein” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to the nearest multiple of 0.1 g; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
13 Amount of potassium “Potassium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
14 Amount of calcium “Calcium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
15 Amount of iron “Iron” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
16 % Daily Value interpretative statement “*5% or less is a little, 15% or more is a lot” [not applicable] The “% Daily Value” or “% DV” subheading is followed by an asterisk in order to reference the % Daily Value interpretative statement shown in the nutrition facts table.
- SOR/2003-11, s. 20
- SOR/2007-176, s. 5
- SOR/2016-305, ss. 18, 73, 75(F)
- SOR/2021-57, s. 9
- SOR/2022-143, s. 18(E)
- SOR/2022-168, s. 14
- SOR/2022-168, s. 52
- SOR/2022-169, s. 15
Additional Information
B.01.402 (1) The nutrition facts table may also contain information set out in column 1 of the table to this section.
(2) If information set out in column 1 of the table to this section is included in the nutrition facts table, it shall be expressed using a description set out in column 2, in the unit set out in column 3 and in the manner set out in column 4.
(2.1) For the purpose of subsection (2), the serving of stated size set out in a nutrition facts table for a prepackaged product, expressed in the metric unit, shall be used as the basis for determining the information appearing in the nutrition facts table in respect of the energy value and nutrient content of the product.
(2.2) The percentage of the daily value for a vitamin or mineral nutrient shown in the nutrition facts table for a prepackaged product in accordance with subsection (2) shall be established on the basis of the amount, by weight, of the vitamin or mineral nutrient per serving of stated size for the product, rounded off in the applicable manner set out in column 4 of the table to this section.
(3) The amount of omega-6 polyunsaturated fatty acids, omega-3 polyunsaturated fatty acids and monounsaturated fatty acids shall be in the nutrition facts table if
(a) the amount of any of those groups of fatty acids or the amount of polyunsaturated fatty acids is in the nutrition facts table or is shown on the label of the prepackaged product or in any advertisement for the product that is made or placed by or on the direction of the manufacturer of the product; or
(b) the amount of any specific fatty acid is shown on the label of the prepackaged product or in any advertisement for the product that is made or placed by or on the direction of the manufacturer of the product.
(4) If the label of a prepackaged product, or any advertisement for the product that is made or placed by or on the direction of the manufacturer of the product, contains a representation, express or implied, that includes information that is set out in column 1 of the table to this section, that information shall also be in the nutrition facts table.
(5) [Repealed, SOR/2016-305, s. 19]
(6) The nutrition facts table shall show the amount of any sugar alcohol, vitamin or mineral nutrient added to the prepackaged product, except in the case of iodide added to salt for table or general household use or fluoride added to prepackaged water or ice.
(7) The nutrition facts table shall show the amount of any vitamin or mineral nutrient that is declared as a component of one of the prepackaged product’s ingredients other than flour.
(8) [Repealed, SOR/2018-108, s. 395]
(9) If information set out in column 1 of the table to this section is included in the nutrition facts table, it shall be shown
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the product may be shown in that language only and is shown on the label in that language.
TABLE
Additional Information
Item Column 1 Column 2 Column 3 Column 4 Information Description Unit Manner of expression 1 Servings per package “Servings per Container”, “(number of units) per Container”, “Servings per Package”, “(number of units) per Package”, “Servings per (naming the package type)”, or “(number of units) per (naming the package type)” The quantity is expressed in number of servings. (1) The quantity is rounded off
(a) if it is less than 2, to the nearest multiple of 1;
(b) if it is between 2 and 5, to the nearest multiple of 0.5; and
(c) if it is more than 5, to the nearest multiple of 1.
(2) If a quantity is rounded off, it shall be preceded by the word “about”.
(3) If the product is of a random weight, the quantity may be declared as “varied”.
2 Energy value “kilojoules” or “kJ” The value is expressed in kilojoules per serving of stated size. The value is rounded off to the nearest multiple of 10 kilojoules. 3 and 4 [Repealed, SOR/2016-305, s. 19] 5 Amount of polyunsaturated fatty acids “Polyunsaturated Fat”, “Polyunsaturated Fatty Acids”, “Polyunsaturated” or “Polyunsaturates” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
6 Amount of omega-6 polyunsaturated fatty acids (1) If the nutrition facts table includes the amount of polyunsaturated fatty acids: “Omega-6”, “Omega-6 Polyunsaturated Fat”, “Omega-6 Polyunsaturated Fatty Acids”, “Omega-6 Polyunsaturates” or “Omega-6 Polyunsaturate
(2) In all other cases: “Omega-6 Polyunsaturated Fat”, “Omega-6 Polyunsaturated Fatty Acids”, “Omega-6 Polyunsaturates” or “Omega-6 Polyunsaturated”
The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
7 Amount of omega-3 polyunsaturated fatty acids (1) If the nutrition facts table includes the amount of polyunsaturated fatty acids: “Omega-3”, “Omega-3 Polyunsaturated Fat”, “Omega-3 Polyunsaturated Fatty Acids”, “Omega-3 Polyunsaturates” or “Omega-3 Polyunsaturated”
(2) In all other cases: “Omega-3 Polyunsaturated Fat”, “Omega-3 Polyunsaturated Fatty Acids”, “Omega-3 Polyunsaturates” or “Omega-3 Polyunsaturated”
The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
8 Amount of monounsaturated fatty acids “Monounsaturated Fat”, “Monounsaturated Fatty Acids”, “Monounsaturates” or “Monounsaturated” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
9 [Repealed, SOR/2016-305, s. 19] 10 Amount of soluble fibre “Soluble Fibre” or “Soluble Fiber” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
11 Amount of insoluble fibre “Insoluble Fibre” or “Insoluble Fiber” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
12 Amount of sugar alcohol (1) If the food contains only one type of sugar alcohol: “Sugar Alcohol”, “Polyol” or “(naming the sugar alcohol)”
(2) In all other cases: “Sugar Alcohols” or “Polyols”
The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
13 Amount of starch “Starch” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
14 Amount of vitamin A “Vitamin A” or “Vit A” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 µg, to “0 µg”;
(b) if it is 5 µg or more but less than 50 µg, to the nearest multiple of 10 µg;
(c) if it is 50 µg or more but less than 250 µg, to the nearest multiple of 50 µg; and
(d) if it is 250 µg or more, to the nearest multiple of 100 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
15 Amount of vitamin C “Vitamin C” or “Vit C” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 mg, to “0 mg”;
(b) if it is 0.1 mg or more but less than 1 mg, to the nearest multiple of 0.2 mg;
(c) if it is 1 mg or more but less than 5 mg, to the nearest multiple of 0.5 mg; and
(d) if it is 5 mg or more, to the nearest multiple of 1 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
16 Amount of vitamin D “Vitamin D” or “Vit D” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 µg, to “0 µg”;
(b) if it is 0.1 µg or more but less than 1 µg, to the nearest multiple of 0.2 µg;
(c) if it is 1 µg or more but less than 5 µg, to the nearest multiple of 0.5 µg; and
(d) if it is 5 µg or more, to the nearest multiple of 1 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
17 Amount of vitamin E “Vitamin E” or “Vit E” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
18 Amount of vitamin K “Vitamin K” or “Vit K” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
19 Amount of thiamine “Thiamine”, “Thiamin”, “Thiamine (Vitamin B1)”, “Thiamine (Vit B1)”, “Thiamin (Vitamin B1)” or “Thiamin (Vit B1)” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
20 Amount of riboflavin “Riboflavin”, “Riboflavin (Vitamin B2)” or “Riboflavin (Vit B2)” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
21 Amount of niacin “Niacin” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
22 Amount of vitamin B6 “Vitamin B6” or “Vit B6” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
23 Amount of folate “Folate” The amount is expressed
(a) in micrograms of dietary folate equivalents (DFE) per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 µg DFE, to “0 µg DFE”;
(b) if it is 1 µg DFE or more but less than 10 µg DFE, to the nearest multiple of 2 µg DFE;
(c) if it is 10 µg DFE or more but less than 50 µg DFE, to the nearest multiple of 5 µg DFE; and
(d) if it is 50 µg DFE or more, to the nearest multiple of 10 µg DFE.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg DFE, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
24 Amount of vitamin B12 “Vitamin B12” or “Vit B12” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 µg, to “0 µg”;
(b) if it is 0.005 µg or more but less than 0.05 µg, to the nearest multiple of 0.01 µg;
(c) if it is 0.05 µg or more but less than 0.25 µg, to the nearest multiple of 0.025 µg; and
(d) if it is 0.25 µg or more, to the nearest multiple of 0.05 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
25 Amount of biotin “Biotin” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
26 Amount of pantothenic acid “Pantothenic Acid” or “Pantothenate” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.01 mg, to “0 mg”;
(b) if it is 0.01 mg or more but less than 0.1 mg, to the nearest multiple of 0.02 mg;
(c) if it is 0.1 mg or more but less than 0.5 mg, to the nearest multiple of 0.05 mg; and
(d) if it is 0.5 mg or more, to the nearest multiple of 0.1 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
27 Amount of choline “Choline” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 mg, to “0 mg”;
(b) if it is 1 mg or more but less than 10 mg, to the nearest multiple of 2 mg;
(c) if it is 10 mg or more but less than 50 mg, to the nearest multiple of 5 mg; and
(d) if it is 50 mg or more, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
28 Amount of phosphorous “Phosphorus” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
29 Amount of iodide “Iodide” or “Iodine” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 µg, to “0 µg”;
(b) if it is 1 µg or more but less than 10 µg, to the nearest multiple of 2 µg;
(c) if it is 10 µg or more but less than 50 µg, to the nearest multiple of 5 µg; and
(d) if it is 50 µg or more, to the nearest multiple of 10 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
30 Amount of magnesium “Magnesium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 mg, to “0 mg”;
(b) if it is 1 mg or more but less than 10 mg, to the nearest multiple of 2 mg;
(c) if it is 10 mg or more but less than 50 mg, to the nearest multiple of 5 mg; and
(d) if it is 50 mg or more, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
31 Amount of zinc “Zinc” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
32 Amount of selenium “Selenium” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 µg, to “0 µg”;
(b) if it is 0.1 µg or more but less than 1 µg, to the nearest multiple of 0.2 µg;
(c) if it is 1 µg or more but less than 5 µg, to the nearest multiple of 0.5 µg; and
(d) if it is 5 µg or more, to the nearest multiple of 1 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
33 Amount of copper “Copper” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.0015 mg, to “0 mg”;
(b) if it is 0.0015 mg or more but less than 0.025 mg, to the nearest multiple of 0.002 mg;
(c) if it is 0.025 mg or more but less than 0.05 mg, to the nearest multiple of 0.005 mg; and
(d) if it is 0.05 mg or more, to the nearest multiple of 0.01 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
34 Amount of manganese “Manganese” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
35 Amount of chromium “Chromium” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
36 Amount of molybdenum “Molybdenum” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 µg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
37 Amount of chloride “Chloride” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as 0 mg, to “0%”; and
(b) in all other cases, to the nearest multiple of 1%.
- SOR/2003-11, s. 20
- err., Vol. 137, No. 5
- SOR/2005-98, s. 2(F)
- SOR/2016-305, ss. 19, 75(F)
- SOR/2018-108, s. 395
Foods for Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2016-305, s. 20
B.01.403 (1) This section applies to a prepackaged product that is intended solely for infants six months of age or older but less than one year of age.
(2) The nutrition facts table of the prepackaged product shall not contain the percentage of the daily value of fat, fibre, sugars, cholesterol or sodium or of the sum of saturated fatty acids and trans fatty acids.
(3) The nutrition facts table of the product may omit the amount of saturated fatty acids, trans fatty acids and cholesterol.
(4) Despite subsection (3), if the amount of cholesterol is in the nutrition facts table, the amounts of saturated fatty acids and trans fatty acids shall also be in the nutrition facts table.
(5) If the information in respect of five or more of the energy value and nutrients referred to in column 1 of items 2, 3 and 8 to 15 of the table to section B.01.401 may be expressed as “0” in the nutrition facts table of the prepackaged product in accordance with that section, the nutrition facts table need only include the following information:
(a) the serving of stated size;
(b) the energy value;
(c) the amount of fat;
(d) the amount of carbohydrate;
(e) the amount of protein;
(f) the amount of any nutrient that is the subject of a representation referred to in subparagraph B.01.401(3)(e)(ii);
(g) the amount of any sugar alcohol, vitamin or mineral nutrient added to the product, other than fluoride added to prepackaged water or ice;
(h) the amount of any vitamin or mineral nutrient that is declared as a component of one of the product’s ingredients other than flour;
(i) the amount of any nutrient referred to in column 1 of any of items 8, 10 or 11 and 13 to 15 of the table to section B.01.401 that may not be expressed as “0” in the nutrition facts table;
(j) except in the case described in paragraph (k), the statement “Not a significant source of (naming each nutrient that is omitted from the nutrition facts table in accordance with this subsection)”, but such a statement may be omitted in respect of saturated fatty acids, trans fatty acids and cholesterol; and
(k) if the product meets the condition specified in subsection B.01.462(3), the statement “Not a significant source of other nutrients” or the statement referred to in paragraph (j).
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 21, 75(F)
Food for Use in Manufacturing Other Foods
B.01.404 (1) Subject to section B.29.004, this section applies to a prepackaged product intended solely for use as an ingredient in the manufacture of other prepackaged products intended for sale to a consumer at the retail level or as an ingredient in the preparation of food by a commercial or industrial enterprise or an institution.
(2) No person shall sell the product unless written nutrition information concerning the product accompanies the product when it is delivered to the purchaser.
(3) The nutrition information
(a) shall include the information that would, but for subsection B.01.401(7), be required by sections B.01.401 and B.01.402 to be included in a nutrition facts table for the product;
(b) may include other information that is permitted by section B.01.402 to be included in that nutrition facts table; and
(c) shall be expressed in accordance with sections B.01.401 and B.01.402, subject to the following modifications, namely,
(i) information for vitamins referred to in subsection D.01.002(1) shall be expressed in the applicable unit referred to in subsection D.01.003(1), and information for mineral nutrients referred to in paragraphs D.02.001(1)(a) to (j), (l) to (n) and (p) shall be expressed in milligrams for sodium, potassium, calcium, phosphorus, magnesium, iron, zinc, chloride, copper and manganese and in micrograms for iodide, chromium, selenium and molybdenum,
(A) per gram or 100 grams of the food, if the net quantity of the food is declared on the label by weight or by count, or
(B) per millilitre or 100 millilitres of the food, if the net quantity of the food is declared on the label by volume,
(ii) information for other nutrients and the energy value set out in column 1 of the table to section B.01.401 or in column 1 of the table to section B.01.402 shall be expressed in the units referred to in column 3,
(A) per gram or 100 grams of the food, if the net quantity of the food is declared on the label by weight or by count, or
(B) per millilitre or 100 millilitres of the food, if the net quantity of the food is declared on the label by volume,
(iii) percentages of daily values and information on servings of stated size may be omitted, and
(iv) the nutrition information shall be stated with a degree of precision that corresponds to the accuracy of the analytical methodology used to produce the information.
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 22, 73, 75(F)
- SOR/2022-169, s. 16
Foods for Enterprise or Institution
B.01.405 (1) This section applies to a prepackaged product, other than a supplemented food, that is a ready-to-serve multiple-serving prepackaged product intended solely to be served in a commercial or industrial enterprise or an institution.
(2) No person shall sell the product unless written nutrition information concerning the product accompanies the product when it is delivered to the purchaser.
(3) The nutrition information
(a) shall include the information that would, but for subsection B.01.401(7), be required by sections B.01.401 and B.01.402 to be included in a nutrition facts table for the product;
(b) may include other information that is permitted by section B.01.402 to be included in that nutrition facts table; and
(c) shall be expressed in accordance with sections B.01.401 and B.01.402.
- SOR/2003-11, s. 20
- SOR/2016-305, s. 23
- SOR/2022-169, s. 17
Basis of Information
B.01.406 (1) Subject to subsections (2) to (8), the information in the nutrition facts table shall be set out only on the basis of the prepackaged product as offered for sale.
(2) If a prepackaged product contains separately packaged ingredients or foods that are intended to be consumed together, the information in the nutrition facts table shall be set out for each ingredient or food or for the entire product.
(3) If a prepackaged product contains an assortment of foods of the same type and the typical serving consists of only one of those foods, the information in the nutrition facts table shall be set out
(a) on the basis of each of the foods contained in the product, if the nutrition information set out in column 1 of the table to section B.01.401 for each of those foods is different; or
(b) on the basis of one of the foods contained in the product, if the nutrition information set out in column 1 of the table to section B.01.401 for each of those foods is the same.
(4) If a prepackaged product contains an assortment of foods of the same type and the typical serving consists of more than one of those foods, the information in the nutrition facts table shall be set out for each of the foods contained in the product or as a composite value.
(5) If a prepackaged product contains a food that is to be prepared in accordance with directions provided in or on the package or that is commonly combined with other ingredients or another food or cooked before being consumed, the nutrition facts table may also set out information for the food as prepared, in which case
(a) the nutrition facts table shall set out the following information for the food as prepared, namely,
(i) except in the case described in subparagraph (ii), the amount of the food expressed using the unit referred to in column 3 of subparagraph 1(b)(i) of the table to section B.01.401 as “about (naming the serving size)” or “about (naming the serving size) prepared” and, if applicable, in the manner specified in column 4 of subitems 1(1) and (2), and
(ii) if the food is commonly served combined with another food, the amount of the other food expressed using the unit referred to in column 3 of subparagraph 1(b)(i) of the table to section B.01.401,
(iii) the energy value, expressed using a description set out in column 2 of item 2 of the table to section B.01.401, in the unit set out in column 3 and in the manner set out in column 4, and
(iv) the information set out in column 1 of items 3, 6 to 8, 11 and 13 to 15 of the table to section B.01.401 and in column 1 of items 14 to 37 of the table to section B.01.402 that is declared as a percentage of the daily value in the nutrition facts table for the food as sold, expressed using a description set out in column 2, as a percentage of the daily value per serving of stated size and in the manner specified in column 4; and
(v) [Repealed, SOR/2016-305, s. 24]
(b) the nutrition facts table may also set out the following information for the added ingredients or the other food, if it is declared in the nutrition facts table for the food as sold, namely,
(i) the information set out in column 1 of items 3 to 5 and 7 to 12 of the table to section B.01.401, expressed using a description set out in column 2, in milligrams for the information set out in column 1 of items 7 and 8 and in grams for the information set out in column 1 of items 3 to 5 and 9 to 12 and in the manner specified in column 4,
(ii) the information set out in column 1 of items 5 to 8 and 10 to 13 of the table to section B.01.402, expressed using a description set out in column 2, in grams and in the manner specified in column 4; and
(iii) the information set out in column 1 of item 2 of the table to section B.01.401, expressed using a description set out in column 2, in the unit set out in column 3 per serving of stated size of the food as prepared, and in the manner specified in column 4.
(6) Subsection (5) does not apply in respect of a prepackaged product that is intended solely for infants six months of age or older but less than one year of age.
(7) Subject to subsection (8), the information in the nutrition facts table may also be set out on the basis of other amounts of a food that reflect different uses or different units of measurement of a food, in which case
(a) the nutrition facts table shall set out the following information for each of the other amounts of food, namely,
(i) the amount of the food expressed in a household measure and a metric measure and in the manner specified in column 4 of subitems 1(1) and (2) of the table to section B.01.401,
(ii) the energy value, expressed using a description set out in column 2 of item 2 of the table to section B.01.401, in the unit set out in column 3 and in the manner set out in column 4, and
(iii) the information set out in column 1 of items 3, 6 to 8, 11 and 13 to 15 of the table to section B.01.401 and in column 1 of items 14 to 37 of the table to section B.01.402 that is declared as a percentage of the daily value in the nutrition facts table for the first amount of food for which information is declared, expressed using a description set out in column 2, as a percentage of the daily value per serving of stated size and in the manner specified in column 4; and
(iv) [Repealed, SOR/2016-305, s. 24]
(b) [Repealed, SOR/2016-305, s. 24]
(c) if the nutrition facts table is set out in a version of the aggregate format specified in section B.01.459 or B.01.464, it shall also set out the following information for each of the other amounts of food, if that information is declared in the nutrition facts table for the first amount of food for which information is declared, namely,
(i) [Repealed, SOR/2016-305, s. 24]
(ii) the information set out in column 1 of items 3 to 5 and 7 to 12 of the table to section B.01.401, expressed using a description set out in column 2, in milligrams for the information set out in column 1 of items 7 and 8 and in grams for the information set out in column 1 of items 3 to 5 and 9 to 12 and in the manner specified in column 4, and
(iii) the information set out in column 1 of items 5 to 8 and 10 to 13 of the table to section B.01.402, expressed using a description set out in column 2, in grams and in the manner specified in column 4.
(8) If the nutrition facts table of a prepackaged product that is intended solely for infants six months of age or older but less than one year of age sets out information in accordance with subsection (7), it shall set out the information referred to in paragraphs (7)(a) and (c).
- SOR/2003-11, s. 20
- SOR/2016-305, s. 24
[
Presentation of Nutrition Facts Table
B.01.450 (1) Subject to subsections (2) to (6), the nutrition facts table shall be presented in accordance with the format specified in the applicable figure in the Directory of NFT Formats, having regard to matters such as order of presentation, dimensions, spacing and the use of upper and lower case letters and bold type.
(2) The characters and rules in the nutrition facts table shall be displayed in a single colour that is a visual equivalent of 100% solid black type on a white background or on a uniform neutral background with a maximum 5% tint of colour.
(3) The characters in the nutrition facts table
(a) shall be displayed in a single standard sans serif font that is not decorative and in such a manner that the characters never touch each other or the rules; and
(b) may be displayed with larger dimensions than those specified in the applicable figure in the Directory of NFT Formats if all the characters in the table are enlarged in a uniform manner.
(3.1) The type size shown in parentheses for a version referred to in a table to sections B.01.454 to B.01.459 or sections B.01.461 to B.01.464 is the minimum type size that may be used in a nutrition facts table to show nutrients set out in the tables to sections B.01.401 and B.01.402 in accordance with that version.
(4) A rule that is specified in the applicable figure in the Directory of NFT Formats as being a 1 point rule or a 2 point rule may be displayed with larger dimensions in the nutrition facts table.
(5) The information in the nutrition facts table shall be in accordance with sections B.01.400 to B.01.403 and B.01.406.
(6) In a nutrition facts table consisting of a table in both English and French, the order of languages may be reversed from the order shown in the applicable figure in the Directory of NFT Formats.
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 25, 74
Location of Nutrition Facts Table
B.01.451 (1) Subject to subsection (2), the nutrition facts table shall be displayed on the label of the prepackaged product
(a) in a table in English and a table in French on the same continuous surface of the available display surface;
(b) in a table in both English and French on a continuous surface of the available display surface; or
(c) in a table in English on a continuous surface of the available display surface and a table in French on another continuous surface of the available display surface that is of the same size and prominence as the first surface.
(2) If in accordance with subsection B.01.012(3) or (7) the information required by these Regulations may be shown on the label of a prepackaged product in English only or in French only and is shown in that language, the nutrition facts table may be displayed on the label of the prepackaged product in a table in that language only on a continuous surface of the available display surface.
- SOR/2003-11, s. 20
Orientation of Nutrition Facts Table
B.01.452 (1) Subject to subsection (2), the nutrition facts table shall be oriented in the same manner as other information appearing on the label of the prepackaged product.
(2) If a version of a nutrition facts table cannot be oriented in the same manner as other information appearing on the label of the prepackaged product, it shall be oriented in another manner if there is sufficient space to do so and the food contained in the package does not leak out and is not damaged when the package is turned over.
(3) Subsection (1) does not apply in respect of a nutrition facts table that is set out on the top or bottom of a prepackaged product.
- SOR/2003-11, s. 20
Application
B.01.453 (1) Sections B.01.454 to B.01.460 apply to prepackaged products other than those that are intended solely for infants six months of age or older but less than one year of age.
(2) Sections B.01.461 to B.01.465 apply to prepackaged products that are intended solely for infants six months of age or older but less than one year of age.
- SOR/2003-11, s. 20
- SOR/2016-305, s. 26
Standard and Horizontal Formats
B.01.454 (1) This section applies to a prepackaged product unless any of sections B.01.455 to B.01.459 applies to the product.
(2) Subject to subsection (3), the nutrition facts table of the prepackaged product shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual standard format in accordance with Figure 3.5(B), 3.6(B) or 3.7(B) of the Directory of NFT Formats;
(b) the bilingual horizontal format in accordance with Figure 4.3(B), 4.4(B) or 4.5(B) of the Directory of NFT Formats;
(c) the linear format in accordance with Figures 16.1(E) and (F) or 16.2(E) and (F) of the Directory of NFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table; or
(e) a manner described in section B.01.466.
(4) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included in that table.
(5) Despite subsections (2) and (3), if the prepackaged product is sold only in the retail establishment where the product is packaged, is labelled by means of a sticker and has an available display surface of 200 cm2 or more, its nutrition facts table shall be set out in a version that is listed in column 1 of items 1 to 3 of Parts 1 to 3 of the table to this section, without regard to any condition specified in column 2.
(6) Despite subsections (2) and (3), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Standard Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 1.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 1.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 1.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 1.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 1.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 1.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Narrow Standard Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 2.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 2.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 2.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 2.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 3
Bilingual Standard Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 3.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 3.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 3.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 3.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 4
Bilingual Horizontal Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 4.1(B) The versions in Parts 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 2 4.2(B) The versions in Parts 1 to 3 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 27, 74
Simplified Formats
B.01.455 (1) This section applies to a prepackaged product if it satisfies the condition set out in subsection B.01.401(6) and its nutrition facts table includes only the information referred to in paragraphs B.01.401(6)(a) to (j).
(2) Subject to subsection (3), the nutrition facts table of the prepackaged product shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table containing only the information referred to in paragraphs B.01.401(6)(a) to (j) in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual simplified standard format in accordance with Figure 6.5(B) or 6.6(B) of the Directory of NFT Formats;
(b) the bilingual simplified horizontal format in accordance with Figure 7.3(B) or 7.4(B) of the Directory of NFT Formats;
(c) the simplified linear format in accordance with Figures 17.1(E) and (F) or 17.2(E) and (F) of the Directory of NFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table; or
(e) a manner described in section B.01.466.
(4) Despite subsections (2) and (3), if the prepackaged product is sold only in the retail establishment where the product is packaged, is labelled by means of a sticker and has an available display surface of 200 cm2 or more, its nutrition facts table shall be set out in a version that is listed in column 1 of items 1 to 3 of Parts 1 and 2 of the table to this section, without regard to any condition specified in column 2.
(5) Despite subsections (2) and (3), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Simplified Standard Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 5.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 5.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 5.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 5.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 5.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 5.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Simplified Standard Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 6.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 6.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 6.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 6.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 3
Bilingual Simplified Horizontal Format
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 7.1(B) The versions in Parts 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 2 7.2(B) The versions in Parts 1 and 2 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 28, 74
Simplified Formats — Single-serving Prepackaged Products
B.01.455.1 (1) This section applies to a single-serving prepackaged product, other than one that is a prepackaged meal, whose nutrition facts table includes only the information referred to in paragraphs B.01.401(6.1)(a) to (j).
(2) Subject to subsection (3), the nutrition facts table of the single-serving prepackaged product shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations, on 15% or less of the available display surface of the single-serving prepackaged product a nutrition facts table containing only the information referred to in paragraphs B.01.401(6.1)(a) to (j) in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual simplified standard format in accordance with Figure 6.5.1(B) or 6.6.1(B) of the Directory of NFT Formats;
(b) the bilingual simplified horizontal format in accordance with Figure 7.3.1(B) or 7.4.1(B) of the Directory of NFT Formats;
(c) the simplified linear format in accordance with Figures 17.1.1(E) and (F) or 17.2.1(E) and (F) of the Directory of NFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table; or
(e) a manner described in section B.01.466.
(4) Despite subsections (2) and (3), if the single-serving prepackaged product is sold only in the retail establishment where the product is packaged, is labelled by means of a sticker and has an available display surface of 200 cm2 or more, its nutrition facts table shall be set out in a version that is listed in column 1 of items 1 to 3 of Part 1 of the table to this section, without regard to any condition specified in column 2.
(5) Despite subsections (2) and (3), if the nutrition facts table of the single-serving prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Bilingual Simplified Standard Format — Single-serving Prepackaged Products
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 6.1.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 6.2.1(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 6.3.1(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 6.4.1(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Simplified Horizontal Format — Single-serving Prepackaged Products
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 7.1.1(B) The versions in Part 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 2 7.2.1(B) The versions in Part 1 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2016-305, s. 29
Dual Format — Foods Requiring Preparation
B.01.456 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product includes information referred to in subsection B.01.406(5), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual dual format in accordance with Figure 9.5(B) or 9.6(B) of the Directory of NFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included in the table, together with the information referred to in subsection B.01.406(5).
(4) Despite subsections (1) and (2), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Dual Format — Foods Requiring Preparation
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 8.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 8.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 8.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 8.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 8.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 8.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Dual Format — Foods Requiring Preparation
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 9.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 9.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 9.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 9.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 30, 74
Aggregate Format — Different Kinds of Foods
B.01.457 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product includes separate information for each food or ingredient as provided in subsection B.01.406(2), paragraph B.01.406(3)(a) or subsection B.01.406(4), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out
(a) in the case of a product described in subsection B.01.406(2) or (4), in
(i) the bilingual aggregate format in accordance with Figure 11.5(B) or 11.6(B) of the Directory of NFT Formats, or
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table, or
(b) in the case of a product described in paragraph B.01.406(3)(a), in
(i) the bilingual aggregate format in accordance with Figure 11.5(B) or 11.6(B) of the Directory of NFT Formats,
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table, or
(iii) a manner described in section B.01.466.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included for each food or ingredient for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in subparagraph (2)(a)(i) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Aggregate Format — Different Kinds of Foods
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 10.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 10.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 10.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 10.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 10.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 10.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Aggregate Format — Different Kinds of Foods
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 11.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 11.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 11.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 11.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 31, 74
Dual Format — Different Amounts of Food
B.01.458 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product includes separate information for different amounts of the food as provided in paragraph B.01.406(7)(a) without including the information referred to in paragraph B.01.406(7)(c), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual dual format in accordance with Figure 13.5(B) or 13.6(B) of the Directory of NFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included for each amount of the food for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Dual Format — Different Amounts of Food
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 12.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 12.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 12.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 12.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 12.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 12.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Dual Format — Different Amounts of Food
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 13.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 13.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 13.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 13.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 32, 74
Aggregate Format — Different Amounts of Food
B.01.459 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product includes separate information for different amounts of the food as provided in paragraphs B.01.406(7)(a) and (c), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual aggregate format in accordance with Figure 15.5(B) or 15.6(B) of the Directory of NFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included for each amount of the food for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the nutrition facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it shall be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Aggregate Format — Different Amounts of Food
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 14.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 14.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 14.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4, 14.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 14.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 14.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Aggregate Format — Different Amounts of Food
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 15.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 15.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 15.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 15.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 33, 74
Presentation of Additional Information
B.01.460 (1) If information referred to in column 1 of the table to section B.01.402 is included in a nutrition facts table that is set out in a version consisting of a table in English and a table in French or a table in English or French, that information shall be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figures 18.1(E) and (F) of the Directory of NFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of NFT Formats.
(2) If information referred to in column 1 of the table to section B.01.402 is included in a nutrition facts table that is set out in a version consisting of a table in both English and French, that information shall be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figure 19.1(B) of the Directory of NFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of NFT Formats.
(3) Despite paragraph (1)(a), the use of indents illustrated in Figures 18.1(E) and (F) of the Directory of NFT Formats is not applicable if information referred to in column 1 of the table to section B.01.402 is set out in the linear format referred to in paragraph B.01.454(3)(c) or the simplified linear format referred to in paragraph B.01.455(3)(c).
- SOR/2003-11, s. 20
- SOR/2016-305, s. 74
Standard and Horizontal Formats — Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2003-11, s. 20
- err.(E), Vol. 137, No. 5
- SOR/2016-305, s. 34
B.01.461 (1) This section applies to a prepackaged product that is intended solely for infants six months of age or older but less than one year of age unless section B.01.462, B.01.463 or B.01.464 applies to the product.
(2) Subject to subsection (3), the nutrition facts table of the prepackaged product shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual standard format in accordance with Figure 22.5(B), 22.6(B) or 22.7(B) of the Directory of NFT Formats;
(b) the bilingual horizontal format in accordance with Figure 23.3(B) or 23.4(B) of the Directory of NFT Formats;
(c) the linear format in accordance with Figures 31.1(E) and (F) or 31.2(E) and (F) of the Directory of NFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table; or
(e) a manner described in section B.01.466.
(4) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included in that table.
TABLE
PART 1
Standard Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 20.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 20.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 20.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 20.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 20.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 20.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Narrow Standard Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 21.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 21.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 21.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 21.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 3
Bilingual Standard Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 22.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 22.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 22.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 22.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 4
Bilingual Horizontal Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 23.1(B) The versions in Parts 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 2 23.2(B) The versions in Parts 1 to 3 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 35, 74
Simplified Formats — Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2016-305, s. 36
B.01.462 (1) This section applies to a prepackaged product that is intended solely for infants six months of age or older but less than one year of age if it satisfies the condition set out in subsection B.01.403(5) and its nutrition facts table includes only the information referred to in paragraphs B.01.403(5)(a) to (k).
(2) Subject to subsection (3), the nutrition facts table of the prepackaged product shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table containing only the information referred to in paragraphs B.01.403(5)(a) to (k) in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual simplified standard format in accordance with Figure 25.5(B) or 25.6(B) of the Directory of NFT Formats;
(b) the bilingual simplified horizontal format in accordance with Figure 26.3(B) or 26.4(B) of the Directory of NFT Formats;
(c) the simplified linear format in accordance with Figures 32.1(E) and (F) or 32.2(E) and (F) of the Directory of NFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table; or
(e) a manner described in section B.01.466.
TABLE
PART 1
Simplified Standard Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 24.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 24.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 24.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 24.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 24.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 24.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Simplified Standard Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 25.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 25.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 25.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 25.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 3
Bilingual Simplified Horizontal Format — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 26.1(B) The versions in Parts 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 2 26.2(B) The versions in Parts 1 and 2 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 37, 74
Aggregate Format — Different Kinds of Foods — Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2016-305, s. 38
B.01.463 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product that is intended solely for infants six months of age or older but less than one year of age includes separate information for each food or ingredient as provided in subsection B.01.406(2), paragraph B.01.406(3)(a) or subsection B.01.406(4), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out
(a) in the case of a product described in subsection B.01.406(2) or (4), in
(i) the bilingual aggregate format in accordance with Figure 28.5(B) or 28.6(B) of the Directory of NFT Formats, or
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table, or
(b) in the case of a product described in paragraph B.01.406(3)(a), in
(i) the bilingual aggregate format in accordance with Figure 28.5(B) or 28.6(B) of the Directory of NFT Formats,
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table, or
(iii) a manner described in section B.01.466.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included for each food or ingredient for which separate information is set out in the table.
TABLE
PART 1
Aggregate Format — Different Kinds of Foods — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 27.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 27.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 27.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 27.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 27.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 27.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Aggregate Format — Different Kinds of Foods — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 28.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 28.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 28.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 28.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 39, 74
Aggregate Format — Different Amounts of Food — Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2016-305, s. 40
B.01.464 (1) Subject to subsection (2), if the nutrition facts table of a prepackaged product that is intended solely for infants six months of age or older but less than one year of age includes separate information for different amounts of the food as provided in subsection B.01.406(8), the nutrition facts table shall be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a nutrition facts table in any of the versions that is listed in column 1 of the table to this section, the nutrition facts table shall be set out in
(a) the bilingual aggregate format in accordance with Figure 30.5(B) or 30.6(B) of the Directory of NFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the nutrition facts table.
(3) For the purpose of this section, in determining whether a version of a nutrition facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the nutrition facts table shall include only the information that is required by these Regulations to be included for each amount of the food for which separate information is set out in the table.
TABLE
PART 1
Aggregate Format — Different Amounts of Food — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 29.1(E) and (F) (nutrients to be shown in a type size of not less than 8 points) 2 29.2(E) and (F) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 29.3(E) and (F) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 29.4(E) and (F) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 5 29.5(E) and (F) The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) 6 29.6(E) and (F) The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed)) PART 2
Bilingual Aggregate Format — Different Amounts of Food — Infants Six Months of Age or Older but Less Than One Year of Age
Item Column 1 Column 2 Figure in Directory of NFT Formats (Version) Condition of use 1 30.1(B) (nutrients to be shown in a type size of not less than 8 points) 2 30.2(B) The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points) 3 30.3(B) The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 7 points (condensed)) 4 30.4(B) The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. (nutrients to be shown in a type size of not less than 6 points (condensed))
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 41, 74
Presentation of Additional Information — Infants Six Months of Age or Older but Less Than One Year of Age
- SOR/2016-305, s. 42
B.01.465 (1) This section applies to a prepackaged product that is intended solely for infants six months of age or older but less than one year of age.
(2) If information referred to in column 1 of the table to section B.01.402 is included in a nutrition facts table that is set out in a version consisting of a table in English and a table in French or a table in English or French, that information shall be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figures 33.1(E) and (F) of the Directory of NFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of NFT Formats.
(3) If information referred to in column 1 of the table to section B.01.402 is included in a nutrition facts table that is set out in a version consisting of a table in both English and French, that information shall be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figure 34.1(B) of the Directory of NFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of NFT Formats.
(4) Despite paragraph (2)(a), the use of indents illustrated in Figures 33.1(E) and (F) of the Directory of NFT Formats is not applicable if information referred to in column 1 of the table to section B.01.402 is set out in the linear format referred to in paragraph B.01.461(3)(c) or the simplified linear format referred to in paragraph B.01.462(3)(c).
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 43, 74
Alternative Methods of Presentation
B.01.466 (1) Despite section A.01.016, the nutrition facts table of a prepackaged product that meets the condition specified in subsection B.01.454(3) or B.01.455(3), paragraph B.01.457(2)(b), subsection B.01.461(3) or B.01.462(3) or paragraph B.01.463(2)(b) may be set out on
(a) a tag attached to the package;
(b) a package insert;
(c) the inner side of a label;
(d) a fold-out label; or
(e) an outer sleeve, overwrap or collar.
(2) If the nutrition facts table is set out in a manner described in paragraph (1)(b) or (c), the outer side of the label of the package shall indicate in a type size of not less than 8 points where the nutrition facts table is located.
(3) If the nutrition facts table is set out in a manner described in subsection (1), it shall be set out
(a) in the case of a product described in subsection B.01.454(3), in a version that is described in paragraph B.01.454(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.01.454;
(b) in the case of a product described in subsection B.01.455(3), in a version that is described in paragraph B.01.455(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.01.455;
(c) in the case of a product described in paragraph B.01.457(2)(b), in a version that is described in subparagraph B.01.457(2)(b)(i) or that is listed in column 1 of the table to section B.01.457;
(d) in the case of a product described in subsection B.01.461(3), in a version that is described in paragraph B.01.461(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.01.461;
(e) in the case of a product described in subsection B.01.462(3), in a version that is described in paragraph B.01.462(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.01.462; and
(f) in the case of a product described in paragraph B.01.463(2)(b), in a version that is described in subparagraph B.01.463(2)(b)(i) or that is listed in column 1 of the table to section B.01.463.
- SOR/2003-11, s. 20
- SOR/2018-69, s. 3(F)
Small Packages
B.01.467 (1) Despite section A.01.016 and subject to subsection (2), if the available display surface of a prepackaged product is less than 100 cm2, the label of the product need not carry a nutrition facts table if the outer side of the label contains an indication of how a purchaser or consumer may obtain the nutrition information that would otherwise be required to be set out in a nutrition facts table on the label of the product.
(2) Subsection (1) does not apply to a prepackaged product that is
(a) described in paragraph B.01.401(3)(a), (b) or (e); or
(b) contained in a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase.
(2.1) However, subsection (1) applies to a prepackaged product that is referred to in subparagraph B.01.401(3)(e)(ii) and that meets the conditions set out in item 37, column 2, of the Table of Permitted Nutrient Content Statements and Claims for the subject “Free of sugars” set out in column 1 if
(a) the product does not contain an added vitamin or mineral nutrient;
(b) the energy value expressed in Calories per serving of stated size and the amount of sugar alcohols expressed in grams per serving of stated size are shown immediately after whichever of the following elements appears last on the label:
(i) the list of ingredients,
(ii) a food allergen source, gluten source and added sulphites statement as defined in subsection B.01.010.1(1),
(iii) a declaration referred to in subsection B.01.010.4(1), or
(iv) any statement referred to in subsection B.01.014(1); and
(c) any statement or claim that is set out in item 37, column 4, of the table for the subject “Free of sugars” set out in column 1 and that appears on the label is
(i) legibly set out on the principal display panel,
(ii) in lower case letters except for the first letter of each word of the statement or claim, which may be an upper case letter,
(iii) of at least the same size and prominence as the letters used in the numerical portion of the declaration of net quantity as required under paragraph 229(1)(a) and subsections 229(2) and (3) of the Safe Food for Canadians Regulations, and
(iv) displayed in a colour contrasting with the background of the label.
(3) An indication referred to in subsection (1)
(a) shall be set out in a type size of not less than 8 points;
(b) shall include a postal address or a toll-free telephone number; and
(c) shall be
(i) in English and French, or
(ii) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the product may be shown in that language only and is shown on the label in that language.
(4) The manufacturer of the prepackaged product shall provide the information referred to in subsection (1) to a purchaser or consumer on request
(a) without charge;
(b) in the following manner, namely,
(i) in the official language in which the information is requested or, if specified by the purchaser or consumer, in both official languages, or
(ii) in one of the official languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the product may be shown in that language only and is shown on the label in that language; and
(c) in the form of a nutrition facts table that is set out
(i) in a format, other than a horizontal format, that is specified in any of sections B.01.454 to B.01.459 or B.01.461 to B.01.464 and that would otherwise be carried on the label of the product in accordance with these Regulations, and
(ii) in a version of that format that is listed in column 1 of item 1 of any Part of the table to the applicable section referred to in subparagraph (i).
(5) In this section, official languages means the English language and the French language.
- SOR/2003-11, s. 20
- SOR/2016-305, s. 44
- SOR/2018-108, s. 400
- SOR/2022-168, s. 15
B.01.468 (1) Subject to subsection (2), if a prepackaged product has an available display surface of less than 100 cm2 and has a nutrition facts table on its label, the nutrition facts table need only include
(a) the serving of stated size;
(b) the energy value and, subject to subsection (2), the amount of nutrients referred to in column 1 of items 2 to 15 of the table to section B.01.401 if the amount shown in the nutrition facts table may not be expressed as “0” according to the manner set out in column 4; and
(c) the amount of any sugar alcohol, vitamin or mineral nutrient added to the prepackaged product.
(2) If the label of a prepackaged product, or any advertisement for the product that is made or placed by or on the direction of the manufacturer of the product, contains a representation, express or implied, that includes information that is set out in column 1 of the table to section B.01.401 or B.01.402, that information shall also be in the nutrition facts table.
- SOR/2016-305, s. 45
B.01.469 Despite subsection B.01.401(1), the label of a prepackaged product that has an available display surface of less than 15 cm2 need not carry a nutrition facts table.
- SOR/2016-305, s. 45
[
Nutrient Content Claims
Interpretation
B.01.500 (1) The following definitions apply in this section and in the Table of Permitted Nutrient Content Statements and Claims.
- combination foods
combination foods means the category of food to which belong foods that contain as ingredients foods from more than one food group, or foods from one or more food groups mixed with foods from the category of other foods, such as pizza or lasagna. (aliments composés)
- food group
food group means one of the following categories of foods:
(a) milk products, and milk product alternatives such as fortified plant-based beverages;
(b) meat, poultry and fish, and alternatives such as legumes, eggs, tofu or peanut butter;
(c) bread and grain products; or
(d) vegetables and fruit. (groupe alimentaire)
- other foods
other foods means the category of food to which belong foods that are not part of any food group, including
(a) foods that are mostly fats, such as butter, margarine, oil or lard;
(b) foods that are mostly sugars, such as jam, honey, syrup or confectionery;
(c) snack foods, such as potato chips or pretzels;
(d) beverages, such as water, tea, coffee or soft drinks; and
(e) herbs, spices and condiments, such as pickles, mustard or ketchup. (autres aliments)
- reference food of the same food group
reference food of the same food group means a food that can be substituted in the diet for the food to which it is compared and that belongs to
(a) the same food group as the food to which it is compared, such as cheese as a reference food for milk, or chicken as a reference food for tofu;
(b) the category of other foods, if the food to which it is compared also belongs to that category, such as pretzels as a reference food for potato chips; or
(c) the category of combination foods, if the food to which it is compared also belongs to that category, such as pizza as a reference food for lasagna. (aliment de référence du même groupe alimentaire)
- similar reference food
similar reference food means a food of the same type as the food to which it is compared and that has not been processed, formulated, reformulated or otherwise modified in a manner that increases or decreases the energy value or the amount of a nutrient that is the subject of the comparison, such as whole milk as a similar reference food for partly skimmed milk or regular chocolate chip cookies as a similar reference food for fat-reduced chocolate chip cookies. (aliment de référence similaire)
(2) The similar reference food referred to in column 3 of item 45 of the Table of Permitted Nutrient Content Statements and Claims, with respect to the subject “light in energy or fat” set out in column 1, shall have a nutrient value that is representative of foods of that type that have not been processed, formulated, reformulated or otherwise modified in a manner that increases the energy value or the amount of fat.
- SOR/2003-11, s. 20
- SOR/2007-302, s. 4(F)
- SOR/2022-168, s. 52
Languages
B.01.501 The representations provided for in sections B.01.503 to B.01.513 that appear on the label of a food shall be
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the food may be shown in that language only and is shown on the label in that language.
- SOR/2003-11, s. 20
Statements or Claims
B.01.502 (1) No person shall, on the label of or in any advertisement for a food, make a representation, express or implied, that characterizes the energy value of the food or the amount of a nutrient contained in the food.
(2) Subsection (1) does not apply to
(a) a representation otherwise provided for in these Regulations, including one that is in the form of a nutrition symbol;
(b) a representation provided for by section 273 of the Safe Food for Canadians Regulations;
(c) a representation provided for by column 1 of Table 2 to Volume 7 of the document entitled Canadian Standards of Identity, prepared by the Canadian Food Inspection Agency and published on its website, as amended from time to time;
(d) a representation that characterizes the amount of lactose in a food;
(e) a representation that characterizes the addition of salt to a food, other than any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims;
(f) a representation that characterizes the addition of sugars to a food, other than any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims;
(g) a representation that characterizes the amount of starch in a food, if the food is intended solely for infants six months of age or older but less than one year of age;
(h) the representations “defatted (naming the food)”, “demineralized (naming the food)” and “high (naming the monosaccharide or disaccharide) (naming the syrup)”;
(i) a representation that characterizes the amount of a fatty acid in a vegetable oil and forms part of its common name;
(j) a representation that characterizes the amount of alcohol in a beverage;
(k) the representation “light salted” with respect to fish; or
(l) the English representation “lean” with respect to a prepackaged meal represented for use in a weight reduction diet or a weight maintenance diet.
B.01.503 (1) A person may, on the label of or in any advertisement for a food, make a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, with respect to a subject set out in column 1, if
(a) the food meets the applicable conditions set out in column 2;
(b) the label or advertisement meets the conditions, if any, set out in column 3, in accordance with sections B.01.504 to B.01.506; and
(c) in the case of a food that is not a prepackaged product, or a prepackaged product for which an advertisement is not made or placed by or on the direction of the manufacturer of the product, the label or advertisement includes, per serving of stated size, and in accordance with section B.01.505 or B.01.506 if applicable,
(i) the declaration of the energy value, if the energy value is the subject of the statement or claim, or
(ii) the amount of the nutrient, if a nutrient is the subject of the statement or claim.
(1.1) Despite subsection (1), no person shall, on the principal display panel of a prepackaged product, make a statement or claim that is set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims and that relates to a nutrient that is referred to in a nutrition symbol that appears on the panel unless it is a statement or claim respecting one of the following subjects set out in column 1:
(a) “reduced in saturated fatty acids”, set out in item 20;
(b) “reduced in sodium or salt”, set out in item 33; or
(c) “reduced in sugars”, set out in item 38.
(2) Despite subsection (1), no person shall, on the label of or in any advertisement for a food that is intended solely for children under four years of age, make a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims unless it is a statement or claim respecting one of the following subjects set out in column 1:
(a) “source of protein”, set out in item 8;
(b) “excellent source of protein”, set out in item 9;
(c) “more protein”, set out in item 10;
(d) “no added sodium or salt”, set out in item 35; or
(e) “no added sugars”, set out in item 40.
(2.01) Despite subsections (1) and (2), it is prohibited, on the label of or in any advertisement for a human milk fortifier, to make a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims.
(2.1) The information required by paragraph (1)(c) need not be included on the label of or in any advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating, if a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims is made on the label or advertisement.
(3) If a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims is made on the label of or in any advertisement for a food, all the words, numbers, signs or symbols that constitute the statement or claim shall be of the same size and prominence.
(4) In the English version of the statements or claims, the word “fibre” may be spelled as “fiber”.
- SOR/2003-11, s. 20
- SOR/2016-305, ss. 46, 75(F)
- SOR/2021-57, s. 10
- SOR/2022-168, s. 17
- SOR/2022-168, s. 52
B.01.504 If a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims is made on the label of a food, the information required under the conditions set out in column 3 shall be
(a) adjacent to, without any intervening printed, written or graphic material,
(i) the statement or claim, if the statement or claim is made only once, or
(ii) the most prominent statement or claim on the principal display panel or, if none appears there, the most prominent statement or claim elsewhere on the label, if the statement or claim is made more than once; and
(b) shown in letters of at least the same size and prominence as
(i) those of the statement or claim, if the statement or claim is made only once, or
(ii) those of the most prominent statement or claim on the principal display panel or, if none appears there, the most prominent statement or claim elsewhere on the label, if the statement or claim is made more than once.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.505 If a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims is made in an advertisement for a food, other than a radio or television advertisement, the information required under the conditions set out in column 3 and, if applicable, the information required by paragraph B.01.503(1)(c), shall be
(a) adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(b) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.506 (1) If a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims is made in a radio or television advertisement, the information required under the conditions set out in column 3 and, if applicable, the information required by paragraph B.01.503(1)(c), shall be provided in the advertisement, except for the information required under the condition set out in paragraph (a) of column 3, in respect of the following subjects set out in column 1, which may be on the label:
(a) “reduced in energy”, set out in item 3;
(b) “reduced in fat”, set out in item 13;
(c) “reduced in saturated fatty acids”, set out in item 20;
(d) “reduced in trans fatty acids”, set out in item 23;
(e) “reduced in cholesterol”, set out in item 29;
(f) “reduced in sodium or salt”, set out in item 33;
(g) “lightly salted”, set out in item 36;
(h) “reduced in sugars”, set out in item 38; and
(i) “light in energy or fat”, set out in item 45.
(2) Despite subsection (1), if the statement or claim is made in a radio or television advertisement that is not made or placed by or on the direction of the manufacturer of the food, the information required under the condition set out in paragraph (a) of column 3 of the Table of Permitted Nutrient Content Statements and Claims, in respect of the subjects set out in paragraphs (1)(a) to (i), shall be provided in the advertisement.
(3) If the information required under the conditions set out in column 3 of the Table of Permitted Nutrient Content Statements and Claims and the information required by paragraph B.01.503(1)(c) is provided in a radio advertisement or in the audio portion of a television advertisement, that information shall immediately precede or follow the statement or claim.
(4) In the case of a television advertisement, the information required under the conditions set out in column 3 of the Table of Permitted Nutrient Content Statements and Claims and, if applicable, the information required by paragraph B.01.503(1)(c), shall be communicated
(a) in the audio mode, if the statement or claim is made only in the audio portion of the advertisement or in both the audio and visual portions; or
(b) in the audio or visual mode, if the statement or claim is made only in the visual portion of the advertisement.
(5) If the information required under the conditions set out in column 3 of the Table of Permitted Nutrient Content Statements and Claims and the information required by paragraph B.01.503(1)(c) is communicated in the visual mode of a television advertisement, it shall
(a) appear concurrently with and for at least the same amount of time as the statement or claim;
(b) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(c) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.507 A person may, on the label of or in any advertisement for a food, make a representation, express or implied, that the food is for use in an energy-reduced diet, if a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, in respect of any of the following subjects set out in column 1, is made on the label of or in the advertisement for the food, in accordance with section B.01.503:
(a) “free of energy”, set out in item 1;
(b) “low in energy”, set out in item 2;
(c) “reduced in energy”, set out in item 3;
(d) “lower in energy”, set out in item 4; or
(e) “free of sugars”, set out in item 37.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.508 (1) A person may, on the label of or in any advertisement for a food, make a representation, express or implied, that the food is for use in a sodium-restricted diet, if a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, in respect of any of the following subjects set out in column 1, is made on the label of or in the advertisement for the food, in accordance with section B.01.503:
(a) “free of sodium or salt”, set out in item 31;
(b) “low in sodium or salt”, set out in item 32;
(c) “reduced in sodium or salt”, set out in item 33; or
(d) “lower in sodium or salt”, set out in item 34.
(2) Despite subsection (1), no person shall, on the principal display panel of a prepackaged product, make a representation, express or implied, that the product is for use in a sodium-restricted diet if a nutrition symbol referring to sodium appears on the panel.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 18
- SOR/2022-168, s. 52
B.01.509 (1) A person may, on the label of or in any advertisement for a food, make the statement or claim that the food is “unsweetened” if
(a) the food meets the conditions set out in item 40, column 2, of the Table of Permitted Nutrient Content Statements and Claims for the subject “No added sugars” set out in column 1; and
(b) the food does not contain a sweetener referred to in section 2 of the Marketing Authorization for Food Additives That May Be Used as Sweeteners.
(2) Despite subsection (1), no person shall, on the principal display panel of a prepackaged product, make a statement or claim that the product is “unsweetened” if a nutrition symbol referring to sugars appears on the panel.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 19
B.01.510 A statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, respecting the following subjects set out in column 1, that is made on the label of or in an advertisement for a breakfast cereal with milk, shall be accompanied by an indication that it refers to 30 g of the breakfast cereal combined with 125 mL of milk:
(a) “source of protein”, set out in item 8;
(b) “excellent source of protein”, set out in item 9; and
(c) “more protein”, set out in item 10.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.511 (1) For greater certainty and subject to subsections (2) to (4), a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims that is made on the label of or in any advertisement for a food may be preceded or followed by other words, numbers, signs or symbols, but none of those shall be interposed between the words, numbers, signs or symbols of the statement or claim.
(2) The words “very”, “ultra” and “extra”, and all other words, numbers, signs or symbols that modify the nature of a statement or claim, shall not precede or follow the statement or claim.
(3) A statement or claim that is made on the label of or in any advertisement for a food that has not been processed, formulated, reformulated or otherwise modified to meet the conditions set out in column 2 of the Table of Permitted Nutrient Content Statements and Claims shall not be accompanied by the brand name of the food.
(4) Any words, numbers, signs or symbols preceding or following the statement or claim referred to in subsection (3) shall accompany the statement or claim in such a manner that the statement or claim characterizes all foods of that type, and not only the specific food.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
B.01.512 If a food meets the conditions set out in column 2 of the Table of Permitted Nutrient Content Statements and Claims for more than one of the subjects set out in column 1, it is not necessary to repeat the common element of the statements or claims set out in column 4 that are used on the label of or in the advertisement for the food, and the remaining elements may be joined by means of a conjunction or punctuation, as appropriate.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 52
Sensory Characteristic
B.01.513 (1) No person shall, on the label of or in any advertisement for a food, make the statement or claim “light” or “léger” — including any phonetic rendering of that statement or claim — respecting a sensory characteristic of the food unless the following conditions are met:
(a) if the statement or claim “light” or “léger” is made on the label of a food, the sensory characteristic shall be
(i) adjacent to, without any intervening printed, written or graphic material,
(A) the statement or claim, if the statement or claim is made only once, or
(B) the most prominent statement or claim on the principal display panel or, if none appears there, the most prominent statement or claim elsewhere on the label, if the statement or claim is made more than once, and
(ii) shown in letters of at least the same size and prominence as
(A) those of the statement or claim, if the statement or claim is made only once, or
(B) those of the most prominent statement or claim on the principal display panel or, if none appears there, the most prominent statement or claim elsewhere on the label, if the statement or claim is made more than once;
(b) if the statement or claim “light” or “léger” is made in an advertisement for a food, other than a radio or television advertisement, the sensory characteristic shall be
(i) adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once, and
(ii) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once;
(c) if the statement or claim “light” or “léger” is made in a radio advertisement or in the audio portion of a television advertisement, the sensory characteristic shall immediately precede or follow the statement or claim; and
(d) if the statement or claim “light” or “léger” is made in the visual portion of a television advertisement, the sensory characteristic shall
(i) appear concurrently with and for the same amount of time as the statement or claim,
(ii) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once, and
(iii) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
(2) Subsection (1) does not apply to the statement or claim “light” or “léger” when it is used with respect to rum.
- SOR/2003-11, s. 20
- err.(F), Vol. 137, No. 5
- SOR/2007-176, s. 6
- SOR/2016-305, s. 75(F)
- SOR/2018-108, s. 397
- SOR/2022-168, s. 20
[
Health Claims
Languages
B.01.600 A statement or claim set out in column 1 of the table following section B.01.603 that appears on the label of a food shall be
(a) in English and French; or
(b) in one of those languages, if in accordance with subsection B.01.012(3) or (7) the information that is required by these Regulations to be shown on the label of the food may be shown in that language only and is shown on the label in that language.
- SOR/2003-11, s. 20
Statements or Claims
B.01.601 (1) A food with a label or advertisement that carries a statement or claim set out in column 1 of the table following section B.01.603 is exempt from the provisions of the Act and its Regulations with respect to drugs, and from subsections 3(1) and (2) of the Act, if
(a) the food meets the applicable conditions set out in column 2;
(b) the label of or the advertisement for the food meets the applicable conditions set out in column 3; and
(c) the food is not
(i) intended solely for children under four years of age, or
(ii) a food represented for use in a very low energy diet.
(2) Subsection (1) does not apply to a food that comes within the definition of drug as defined in section 2 of the Act for a reason other than the fact that its label or advertisement carries a statement or claim referred to in that subsection.
(3) Subsection (1) applies even if the word “graisses” in the French version of the statement or claim is replaced by the word “lipides”.
- SOR/2003-11, s. 20
- SOR/2022-168, s. 21
B.01.602 (1) The information required under the conditions set out in column 3 of the table following section B.01.603 that appears in an advertisement for a food that is not a prepackaged product, or in an advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, shall,
(a) in the case of an advertisement, other than a radio or television advertisement, be
(i) adjacent to, without any intervening printed, written or graphic material, the statement or claim set out in column 1, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once, and
(ii) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once;
(b) in the case of a radio advertisement or the audio portion of a television advertisement, immediately precede or follow the statement or claim set out in column 1; or
(c) in the case of a television advertisement, be communicated
(i) in the audio mode, if the statement or claim set out in column 1 is made only in the audio portion of the advertisement or in both the audio and visual portions, or
(ii) in the audio or visual mode, if the statement or claim set out in column 1 is made only in the visual portion of the advertisement.
(2) The information that is communicated in the visual mode of a television advertisement in accordance with subparagraph (1)(c)(ii) shall
(a) appear concurrently with and for at least the same amount of time as the statement or claim;
(b) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(c) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
- SOR/2003-11, s. 20
B.01.603 (1) For greater certainty, a statement or claim set out in column 1 of the table following this section that is made on the label of or in any advertisement for a food may be preceded or followed by other words, numbers, signs or symbols, but none of those shall be interposed between the words, numbers, signs or symbols of the statement or claim.
(2) If a food meets the conditions set out in column 2 of the table following this section for more than one item of that table, it is not necessary to repeat the common element of the statements or claims set out in column 1 that are used on the label of or in the advertisement for the food, and the remaining elements may be joined by means of a conjunction or punctuation, as appropriate.
TABLE
Item Column 1 Column 2 Column 3 Statement or Claim Conditions — Food Conditions — Label or Advertisement 1 (1) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is sodium-free.”
(2) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is low in sodium.”
(3) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is a good source of potassium and is sodium-free.”
(4) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is a good source of potassium and is low in sodium.”
(5) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is high in potassium and is sodium-free.”
(6) “A healthy diet containing foods high in potassium and low in sodium may reduce the risk of high blood pressure, a risk factor for stroke and heart disease. (Naming the food) is high in potassium and is low in sodium.”
The food
(a) other than a vegetable or fruit, does not meet the conditions set out in column 2 of item 2 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in energy” set out in column 1;
(b) contains at least 10% of the weighted recommended nutrient intake of a vitamin or a mineral nutrient
(i) per reference amount and per serving of stated size, or
(ii) per serving of stated size, if the food is a prepackaged meal;
(c) meets the conditions set out in column 2 of item 19 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in saturated fatty acids” set out in column 1;
(d) contains 0.5% or less alcohol;
(e) meets the conditions set out in column 2 of item 31 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of sodium or salt” set out in column 1, if the label of or advertisement for the food carries statement or claim (1), (3) or (5) set out in column 1 of this item;
(f) meets the conditions set out in column 2 of item 32 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in sodium or salt” set out in column 1, if the label of or advertisement for the food carries statement or claim (2), (4) or (6) set out in column 1 of this item; and
(g) contains 350 mg or more of potassium, if the label of or advertisement for the food carries statement or claim (3), (4), (5) or (6) set out in column 1 of this item,
(i) per reference amount and per serving of stated size, or
(ii) per serving of stated size, if the food is a prepackaged meal.
If the statement or claim is made on the label of or in the advertisement for a food that is not a prepackaged product, or in the advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the label or advertisement shall include the amount of sodium and potassium per serving of stated size, in accordance with section B.01.602, if applicable. However, this requirement does not apply if the statement or claim is made on the label of or in the advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
2 (1) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is a good source of calcium.”
(2) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is high in calcium.”
(3) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is an excellent source of calcium.”
(4) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is very high in calcium.”
(5) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is an excellent source of calcium and vitamin D.”
(6) “A healthy diet with adequate calcium and vitamin D, and regular physical activity, help to achieve strong bones and may reduce the risk of osteoporosis. (Naming the food) is very high in calcium and vitamin D.”
The food
(a) other than a vegetable or fruit, does not meet the conditions set out in column 2 of item 2 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in energy” set out in column 1;
(b) contains no more phosphorus, excluding that provided by phytate, than calcium;
(c) contains 0.5% or less alcohol;
(d) contains, if the label of or advertisement for the food carries statement or claim (1) or (2) set out in column 1 of this item,
(i) 200 mg or more of calcium per reference amount and per serving of stated size, or
(ii) 300 mg or more of calcium per serving of stated size, if the food is a prepackaged meal;
(e) contains, if the label of or advertisement for the food carries statement or claim (3), (4), (5) or (6) set out in column 1 of this item,
(i) 275 mg or more of calcium per reference amount and per serving of stated size, or
(ii) 400 mg or more of calcium per serving of stated size, if the food is a prepackaged meal; and
(f) contains 1.25 µg or more of vitamin D, if the label of or advertisement for the food carries statement or claim (5) or (6) set out in column 1 of this item,
(i) per reference amount and per serving of stated size, or
(ii) per serving of stated size, if the food is a prepackaged meal.
(1) If the statement or claim is made on the label of a prepackaged product, or in any advertisement for the prepackaged product that is made or placed by or on the direction of the manufacturer of the product, the amount of vitamin D and phosphorus shall be included in, as the case may be,
(a) the nutrition facts table in accordance with subsection B.01.402(2); or
(b) the supplemented food facts table in accordance with subsection B.29.002(1), in respect of the description referred to in column 2, the unit referred to in column 3 and the manner of expression referred to in column 4 of the table to section B.29.002, or with subsection B.29.003(3), or with both, as the case may be.
(2) If the statement or claim is made on the label of or in the advertisement for a food that is not a prepackaged product, or in the advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the label or advertisement shall include the amount of vitamin D, calcium and phosphorus per serving of stated size, in accordance with section B.01.602, if applicable. However, this requirement does not apply if the statement or claim is made on the label of or in the advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
3 (1) “A healthy diet low in saturated and trans fats may reduce the risk of heart disease. (Naming the food) is free of saturated and trans fats.”
(2) “A healthy diet low in saturated and trans fats may reduce the risk of heart disease. (Naming the food) is low in saturated and trans fats.”
The food
(a) other than a vegetable or fruit, does not meet the conditions set out in column 2 of item 2 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in energy” set out in column 1;
(b) contains at least 10% of the weighted recommended nutrient intake of a vitamin or a mineral nutrient
(i) per reference amount and per serving of stated size, or
(ii) per serving of stated size, if the food is a prepackaged meal;
(c) contains 100 mg or less of cholesterol per 100 g of food;
(d) contains 0.5% or less alcohol;
(e) if it is a fat or an oil, meets the conditions set out in
(i) column 2 of item 25 of the Table of Permitted Nutrient Content Statements and Claims for the subject “source of omega-3 polyunsaturated fatty acids” set out in column 1,
(ii) column 2 of item 26 of the Table of Permitted Nutrient Content Statements and Claims for the subject “source of omega-6 polyunsaturated fatty acids” set out in column 1, or
(iii) subparagraphs (i) and (ii);
(f) contains
(i) 480 mg or less of sodium per reference amount and per serving of stated size, and per 50 g if the reference amount is 30 g or 30 mL or less, or
(ii) 960 mg or less of sodium per serving of stated size, if the food is a prepackaged meal;
(g) meets the conditions set out in column 2 of item 18 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of saturated fatty acids” set out in column 1, if the label of or advertisement for the food carries statement or claim (1) set out in column 1 of this item; and
(h) meets the conditions set out in column 2 of item 19 of the Table of Permitted Nutrient Content Statements and Claims for the subject “low in saturated fatty acids” set out in column 1, if the label of or advertisement for the food carries statement or claim (2) set out in column 1 of this item.
If the statement or claim is made on the label of or in the advertisement for a food that is not a prepackaged product, or in the advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the label or advertisement shall include the amount of saturated fatty acids and trans fatty acids per serving of stated size, in accordance with section B.01.602, if applicable. However, this requirement does not apply if the statement or claim is made on the label of or in the advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
4 “A healthy diet rich in a variety of vegetables and fruit may help reduce the risk of some types of cancer.”
The food
(a) is one of the following vegetables, fruits or juices and may contain only food additives that are referred to in section 2 of a marketing authorization, sweetening agents, salt, herbs, spices, seasonings and water:
(i) a fresh, frozen, canned or dried vegetable,
(ii) a fresh, frozen, canned or dried fruit,
(iii) a vegetable or fruit juice, or
(iv) a combination of the foods set out in subparagraphs (i) to (iii);
(b) is not one of the following:
(i) potatoes, yams, cassava, plantain, corn, mushrooms, mature legumes and their juices,
(ii) vegetables or fruit used as condiments, garnishes or flavourings, including maraschino cherries, glacé fruit, candied fruit and onion flakes,
(iii) jams or jam-type spreads, marmalades, preserves and jellies,
(iv) olives, or
(v) powdered vegetables or fruit; and
(c) contains 0.5% or less alcohol.
4.1 “A healthy diet rich in a variety of vegetables and fruit may help reduce the risk of heart disease.”
The food
(a) is one of the following vegetables or fruits and may contain only food additives that are referred to in section 2 of a marketing authorization, salt, herbs, spices, seasonings and water:
(i) a fresh, frozen, canned or dried vegetable,
(ii) a fresh, frozen, canned or dried fruit,
(iii) a vegetable juice or vegetable drink, or
(iv) a combination of the foods set out in any of subparagraphs (i) to (iii);
(b) is not one of or does not contain any of the following:
(i) potatoes, yams, cassava, plantain, mature legumes and their juices,
(ii) vegetables or fruit used as condiments, garnishes or flavourings, including maraschino cherries, glacé fruit, candied fruit and onion flakes,
(iii) jams or jam-type spreads, marmalades, preserves and jellies,
(iv) olives,
(v) a fruit juice or fruit drink,
(vi) powdered vegetables or fruit, or
(vii) the seed of a fruit known as a drupe, including almonds, cashews and coconuts;
(c) contains 0.5% or less alcohol; and
(d) contains less than 15% of the daily value of sodium per reference amount and per serving of stated size, and per 50 g if the reference amount is 30 g or 30 mL or less.
5 (1) “Won’t cause cavities.”
(2) “Does not promote tooth decay.”
(3) “Does not promote dental caries.”
(4) “Non-cariogenic.”
The food is a chewing gum, hard candy or breath freshener product that
(a) contains 0.25% or less starch, dextrins, mono-, di- and oligosaccharides or other fermentable carbohydrates combined; or
(b) does not, if it contains more than 0.25% fermentable carbohydrates, lower plaque pH below 5.7 by bacterial fermentation during 30 minutes after consumption as measured by the indwelling plaque pH test, referred to in “Identification of Low Caries Risk Dietary Components” by T.N. Imfeld, Volume 11, Monographs in Oral Science, 1983.
If the statement or claim is made on the label of a prepackaged product, or in any advertisement for the prepackaged product that is made or placed by or on the direction of the manufacturer of the product, the amount of sugar alcohols, if present, shall be included in, as the case may be,
(a) the nutrition facts table, in accordance with subsection B.01.402(2); or
(b) the supplemented food facts table, in accordance with subsection B.29.003(3).
- SOR/2003-11, s. 20
- err.(F), Vol. 137, No. 5
- SOR/2010-142, s. 2
- SOR/2016-305, ss. 47, 48, 75(F)
- SOR/2022-168, s. 52
- SOR/2022-169, s. 18
DIVISION 2Alcoholic Beverages
B.02.001 The foods referred to in this Division are included in the term alcoholic beverage.
- SOR/93-145, s. 3(F)
B.02.002 In this Division,
- absolute alcohol
absolute alcohol means alcohol of a strength of 100 per cent; (alcool absolu)
- age
age means the period during which an alcoholic beverage is kept under such conditions of storage as may be necessary to develop its characteristic flavour and bouquet; (âge)
- alcohol
alcohol means ethyl alcohol; (alcool)
- flavouring
flavouring means, in respect of a spirit, any other spirit or wine, domestic or imported, added as a flavouring to that spirit as authorized under the Excise Act; (substance aromatique)
- grain spirit
grain spirit means an alcoholic distillate, obtained from a mash of cereal grain or cereal grain products saccharified by the diastase of malt or by other enzymes and fermented by the action of yeast or a mixture of yeast and other micro-organisms, and from which all or nearly all of the naturally occurring substances other than alcohol and water have been removed; (esprit de grain)
- malt spirit
malt spirit means an alcoholic distillate, obtained by pot-still distillation from a mash of cereal grain or cereal grain products saccharified by the diastase of malt and fermented by the action of yeast or a mixture of yeast and other micro-organisms; (esprit de malt)
- molasses spirit
molasses spirit means an alcoholic distillate, obtained from sugar-cane or sugar-cane products fermented by the action of yeast or a mixture of yeast and other micro-organisms, from which all or nearly all of the naturally occurring substances other than alcohol and water have been removed; (esprit de mélasse)
- small wood
small wood means wood casks or barrels of not greater than 700 L capacity; (petit fût)
- sweetening agent
sweetening agent means glucose-fructose, fructose syrup or any food for which a standard is provided in Division 18, or any combination thereof. (agent édulcorant)
- SOR/84-300, s. 10
- SOR/93-145, s. 4
B.02.003 If an alcoholic beverage contains 1.1% or more alcohol by volume, the percentage by volume of alcohol present in the alcoholic beverage shall be shown on the principal display panel
(a) followed by the words “alcohol by volume” or the abbreviation “alc./vol.” or “alc/vol”; or
(b) preceded by the abbreviation “alc.” or “alc” and followed by the abbreviation “vol.” or “vol”.
- SOR/88-418, s. 1
- SOR/93-145, s. 5(F)
- SOR/2014-9, s. 1
B.02.004 (1) No person shall sell flavoured purified alcohol in a container with a capacity of 1,000 mL or less unless it contains 25.6 mL or less of alcohol.
(2) Subsection (1) does not apply to flavoured purified alcohol that is sold in a glass container with a capacity of at least 750 mL.
(3) For the purposes of this section, flavoured purified alcohol means an alcoholic beverage
(a) that is obtained from an alcohol base that has been purified during the course of manufacture through a process other than distillation and from which most of the naturally occurring substances other than alcohol and water have been removed; and
(b) to which has been added during the course of manufacture any substance or mixture of substances that imparts flavour.
Whisky
B.02.010 [S]. Whisky or Whiskey, other than Malt Whisky, Scotch Whisky, Irish Whisky, Canadian Whisky, Canadian Rye Whisky, Rye Whisky, Highland Whisky, Bourbon Whisky and Tennessee Whisky,
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained from a mash of cereal grain or cereal grain products saccharified by the diastase of malt or by other enzymes and fermented by the action of yeast or a mixture of yeast and other micro-organisms; and
(b) may contain caramel and flavouring.
- SOR/93-145, s. 6
- SOR/93-603, s. 2
B.02.011 and B.02.012 [Repealed, SOR/93-145, s. 7]
B.02.013 [S]. Malt Whisky
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of a mash of malted grain fermented by the action of yeast or a mixture of yeast and other micro-organisms;
(b) shall possess the aroma, taste and character generally attributed to malt whisky; and
(c) may contain caramel and flavouring.
- SOR/93-145, s. 8
B.02.014 and B.02.015 [Repealed, SOR/93-145, s. 9]
B.02.016 [S]. Scotch Whisky shall be whisky distilled in Scotland as Scotch whisky for domestic consumption in accordance with the laws of the United Kingdom.
B.02.017 No person shall blend or modify in any manner any Scotch whisky that is imported in bulk for the purpose of bottling and sale in Canada as Scotch whisky except by
(a) blending with other Scotch whisky,
(b) the addition of distilled or otherwise purified water to adjust to a required strength, or
(c) the addition of caramel.
B.02.018 [S]. Irish Whisky shall be whisky distilled in Northern Ireland or in the Republic of Ireland as Irish whisky for domestic consumption in accordance with the laws of Northern Ireland or the Republic of Ireland.
B.02.019 No person shall blend or modify in any manner any Irish whisky that is imported in bulk for the purpose of bottling and sale in Canada as Irish whisky except by
(a) blending with other Irish whisky,
(b) the addition of distilled or otherwise purified water to adjust to a required strength, or
(c) the addition of caramel.
B.02.020 [S]. (1) Canadian Whisky, Canadian Rye Whisky or Rye Whisky
(a) shall
(i) be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained from a mash of cereal grain or cereal grain products saccharified by the diastase of malt or by other enzymes and fermented by the action of yeast or a mixture of yeast and other micro-organisms,
(ii) be aged in small wood for not less than three years,
(iii) possess the aroma, taste and character generally attributed to Canadian whisky,
(iv) be manufactured in accordance with the requirements of the Excise Act and the regulations made thereunder,
(v) be mashed, distilled and aged in Canada, and
(vi) contain not less than 40 per cent alcohol by volume; and
(b) may contain caramel and flavouring.
(2) Subject to subsection (3), no person shall make any claim with respect to the age of Canadian whisky, other than for the period during which the whisky has been held in small wood.
(3) Where Canadian whisky has been aged in small wood for a period of at least three years, any period not exceeding six months during which that whisky was held in other containers may be claimed as age.
- SOR/93-145, s. 10
- SOR/2000-51, s. 1
B.02.021 [S]. Highland Whisky
(a) shall be a potable alcoholic beverage blended in Canada from
(i) not less than 25 per cent malt whisky calculated on an absolute alcohol basis, distilled in Canada or Scotland, and
(ii) whisky; and
(b) may, if it contains 51 per cent or more malt whisky distilled in Scotland, be labelled or advertised as containing malt whisky distilled in Scotland.
- SOR/93-145, s. 10
B.02.022 (1) Subject to subsection (2), no person shall label, package, sell or advertise any food as Bourbon Whisky, or in such a manner that it is likely to be mistaken for Bourbon whisky unless it is whisky manufactured in the United States as Bourbon whisky in accordance with the laws of the United States applicable in respect of Bourbon whisky for consumption in the United States.
(2) A person may modify Bourbon whisky that is imported for the purpose of bottling and sale in Canada as Bourbon whisky by the addition of distilled or otherwise purified water to adjust the Bourbon whisky to a required strength.
- SOR/89-59, s. 2
- SOR/93-145, s. 11(F)
B.02.022.1 (1) Subject to subsection (2), no person shall label, package, sell or advertise any food as Tennessee Whisky, or in such a manner that it is likely to be mistaken for Tennessee whisky unless it is a straight Bourbon whisky produced in the State of Tennessee and manufactured in the United States as Tennessee whisky in accordance with the laws of the United States applicable in respect of Tennessee whisky for consumption in the United States.
(2) A person may modify Tennessee whisky that is imported for the purpose of bottling and sale in Canada as Tennessee whisky by the addition of distilled or otherwise purified water to adjust the Tennessee whisky to a required strength.
- SOR/93-603, s. 3
B.02.023 (1) Subject to sections B.02.022 and B.02.022.1, no person shall sell for consumption in Canada any whisky that has not been aged for a period of at least three years in small wood.
(2) Nothing in subsection (1) applies in respect of flavouring contained in whisky, but no person shall sell for consumption in Canada whisky containing any flavouring, other than wine, that has not been aged for a period of at least two years in small wood.
- SOR/93-145, s. 12
- SOR/93-603, s. 4
Rum
B.02.030 [S]. Rum
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained from sugar-cane or sugar-cane products fermented by the action of yeast or a mixture of yeast and other micro-organisms;
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations; and
(c) if it is imported in bulk for the purpose of bottling and sale in Canada as imported rum, may only be
(i) modified by adding distilled or otherwise purified water to adjust the rum to the strength stated on the label applied to the container,
(ii) modified by adding caramel, or
(iii) blended with other imported rum or, in the case of rum sold as Caribbean rum, with other rum.
- SOR/93-145, s. 13
- SOR/2012-292, s. 1
B.02.031 (1) No person shall sell for consumption in Canada any rum that has not been aged for a period of at least one year in small wood.
(2) Nothing in subsection (1) applies in respect of flavouring contained in rum, but no person shall sell for consumption in Canada rum containing any flavouring, other than wine, that has not been aged for a period of at least one year in small wood.
- SOR/84-657, s. 1
- SOR/93-145, s. 13
B.02.032 [Repealed, SOR/93-145, s. 14]
B.02.033 [Repealed, SOR/2012-292, s. 2]
B.02.034 [Repealed, SOR/2012-292, s. 2]
Gin
B.02.040 [S]. Hollands, Hollands Gin, Geneva, Geneva Gin, Genever, Genever Gin or Dutch-type Gin
(a) shall be a potable alcoholic beverage obtained
(i) by the redistillation of malt spirit with or over juniper berries, or by a mixture of the products of more than one such redistillation,
(ii) by the redistillation of a combination of malt spirit and not more than four times its volume on an absolute alcohol basis of grain spirit with or over juniper berries, or by a mixture of the products of more than one such redistillation, or
(iii) by the blending of malt spirit, redistilled with or over juniper berries, with not more than four times its volume on an absolute alcohol basis of grain spirit or molasses spirit, or by a mixture of the products of more than one such blending;
(b) may contain
(i) other aromatic botanical substances, added during the redistillation process, and
(ii) caramel;
(c) shall not contain more than two per cent sweetening agent;
(d) may be labelled or advertised as being distilled, where subparagraph (a)(i) or (ii) is complied with; and
(e) shall be described on the principal display panel of its label and in any advertisements as blended gin, where subparagraph (a)(iii) is complied with.
- SOR/93-145, s. 15
B.02.041 [S]. Gin, other than Hollands, Hollands Gin, Geneva, Geneva Gin, Genever, Genever Gin or Dutch-type Gin,
(a) shall be a potable alcoholic beverage obtained
(i) by the redistillation of alcohol from food sources with or over juniper berries, or by a mixture of the products of more than one such redistillation, or
(ii) by the blending of alcohol from food sources, redistilled with or over juniper berries, with alcohol from food sources or by a mixture of the products of more than one such blending;
(b) may contain
(i) other aromatic botanical substances, added during the redistillation process,
(ii) a sweetening agent, and
(iii) a flavouring preparation for the purpose of maintaining a uniform flavour profile; and
(c) may be labelled or advertised as Dry Gin or London Dry Gin if sweetening agents have not been added.
- SOR/93-145, s. 15
B.02.042 [Repealed, SOR/93-145, s. 15]
B.02.043 No person shall make any claim for age for gin but gin that has been held in suitable containers may bear a label declaration to that effect.
Brandy
B.02.050 [S]. Brandy, other than Armagnac Brandy or Armagnac, Canadian Brandy, Cognac Brandy or Cognac, Dried Fruit Brandy, Fruit Brandy, Grappa, Lees Brandy and Pomace or Marc,
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of wine; and
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations.
- SOR/84-300, s. 12
- SOR/93-145, s. 16
B.02.051 [S]. Armagnac Brandy or Armagnac shall be brandy manufactured in the Armagnac district of France in accordance with the laws of the French Republic for consumption in that country.
- SOR/93-145, s. 16
B.02.052 [S]. Canadian Brandy
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of wine that has been fermented in Canada; and
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations.
- SOR/93-145, s. 16
B.02.053 [S]. Cognac Brandy or Cognac shall be brandy manufactured in the Cognac district of France in accordance with the laws of the French Republic for consumption in that country.
- SOR/93-145, s. 16
B.02.054 [S]. Dried Fruit Brandy
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained from sound dried fruit; and
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations.
- SOR/93-145, s. 16
B.02.055 [S]. Fruit Brandy
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of
(i) fruit wine or a mixture of fruit wines, or
(ii) a fermented mash of sound ripe fruit other than grapes, or a mixture of sound ripe fruits other than grapes;
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations; and
(c) may be described on its label as “(naming the fruit) brandy” if all of the fruit or fruit wine used to make the brandy originates from the named fruit.
- SOR/93-145, s. 16
B.02.056 [S]. Grappa
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of the pomace from sound ripe grapes after the removal of the juice or wine; and
(b) may contain
(i) caramel
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations.
- SOR/93-145, s. 16
B.02.057 [S]. Lees Brandy
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of the lees of wine or fruit wine;
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations; and
(c) may be described on its label as “(naming the fruit) Lees Brandy” if all of the lees used to make the brandy originate from the named fruit.
- SOR/93-145, s. 16
B.02.058 [S]. Pomace or Marc
(a) shall be a potable alcoholic distillate, or a mixture of potable alcoholic distillates, obtained by the distillation of the skin and pulp of sound ripe fruit after the removal of the fruit juice, wine or fruit wine;
(b) may contain
(i) caramel,
(ii) fruit and other botanical substances, and
(iii) flavouring and flavouring preparations; and
(c) may be described on its label as “(naming the fruit) Pomace” or “(naming the fruit) Marc” if all of the skin and pulp used to make the brandy originate from the named fruit.
- SOR/93-145, s. 16
B.02.059 No person shall blend or modify in any manner any brandy that is imported in bulk for the purpose of bottling and sale in Canada as imported brandy, except by
(a) blending with other imported brandy;
(b) the addition of caramel; and
(c) the addition of distilled or otherwise purified water to adjust the brandy to a required strength.
- SOR/93-145, s. 16
B.02.060 Where brandy is wholly distilled in a country other than Canada, the label shall indicate the country of origin.
- SOR/84-300, s. 13(F)
- SOR/93-145, s. 16
B.02.061 (1) No person shall sell any brandy that has not been aged for a period of at least one year in wooden containers or at least six months in small wood.
(2) Nothing in subsection (1) applies in respect of flavouring contained in brandy, but no person shall sell brandy containing any flavouring, other than wine, that has not been aged for a period of at least one year in wooden containers or at least six months in small wood.
(3) Nothing in subsection (1) or (2) applies in respect of brandy that meets the standards prescribed by any of sections B.02.051 to B.02.058.
(4) No person shall make any claim with respect to the age of brandy other than for the period during which the brandy has been held in wooden containers or in small wood.
- SOR/93-145, s. 16
Liqueurs and Spirituous Cordials
B.02.070 [S]. Liqueur or Spirituous Cordial
(a) shall be a product obtained by the mixing or distillation of alcohol from food sources with or over fruits, flowers, leaves or other botanical substances or their juices or with extracts derived by the infusion, percolation or maceration of those botanical substances;
(b) shall have added, during the course of manufacture, a sweetening agent in an amount that is not less than 2.5 per cent of the finished product;
(c) shall contain not less than 23 per cent absolute alcohol by volume; and
(d) may contain
(i) natural and artificial flavouring preparations, and
(ii) colour.
- SOR/93-145, s. 16
Vodka
B.02.080 [S]. (1) Vodka shall be a potable alcoholic distillate obtained from potatoes, cereal grain or any other material of agricultural origin fermented by the action of yeast or a mixture of yeast and other micro-organisms.
(2) The distillate shall be treated with charcoal or other means so that the vodka is without distinctive character, aroma or taste.
(3) Vodka produced, in whole or in part, from material of agricultural origin other than potatoes or cereal grain, shall carry in close proximity to the common name, the statement “produced from” followed by the name of all material of agricultural origin used.
- SOR/93-145, s. 16
- SOR/2019-217, s. 1
Tequila
B.02.090 (1) Subject to subsection (2), no person shall label, package, sell or advertise any food as Tequila, or in such a manner that it is likely to be mistaken for Tequila unless it is Tequila manufactured in Mexico as Tequila in accordance with the laws of Mexico applicable in respect of Tequila for consumption in Mexico.
(2) A person may modify Tequila that is imported for the purpose of bottling and sale in Canada as Tequila by the addition of distilled or otherwise purified water to adjust the Tequila to a required strength.
- SOR/93-603, s. 5
Mezcal
B.02.091 (1) Subject to subsection (2), no person shall label, package, sell or advertise any food as Mezcal, or in such a manner that it is likely to be mistaken for Mezcal unless it is Mezcal manufactured in Mexico as Mezcal in accordance with the laws of Mexico applicable in respect of Mezcal for consumption in Mexico.
(2) A person may modify Mezcal that is imported for the purpose of bottling and sale in Canada as Mezcal by the addition of distilled or otherwise purified water to adjust the Mezcal to a required strength.
- SOR/93-603, s. 6
Wine
B.02.100 [S]. Wine
(a) shall be an alcoholic beverage that is produced by the complete or partial alcoholic fermentation of fresh grapes, grape must, products derived solely from fresh grapes, or any combination of them;
(b) may have added to it during the course of the manufacture
(i) yeast,
(ii) concentrated grape juice,
(iii) dextrose, fructose, glucose or glucose solids, invert sugar, sugar, or aqueous solutions of any of them,
(iv) yeast foods, in accordance with Table XIV to section B.16.100,
(v) calcium sulphate in such quantity that the content of soluble sulphates in the finished wine shall not exceed 0.2 per cent weight by volume calculated as potassium sulphate,
(vi) calcium carbonate in such quantity that the content of tartaric acid in the finished wine shall not be less than 0.15 per cent weight by volume,
(vii) sulphurous acid, including salts thereof, in such quantity that its content in the finished wine shall not exceed
(A) 70 parts per million in the free state, or
(B) 350 parts per million in the combined state, calculated as sulphur dioxide,
(viii) any of the following substances:
(A) citric acid, fumaric acid, lactic acid, malic acid, potassium bicarbonate, potassium carbonate, potassium citrate and tartaric acid, at a maximum level of use consistent with good manufacturing practice,
(B) metatartaric acid at a maximum level of use of 0.01 per cent, and
(C) potassium acid tartrate at a maximum level of use of 0.42 per cent,
(ix) amylase and pectinase at a maximum level of use consistent with good manufacturing practice,
(x) ascorbic acid or erythorbic acid, or their salts, at a maximum level of use consistent with good manufacturing practice,
(xi) antifoaming agents, in accordance with Table VIII to section B.16.100,
(xii) any of the following fining agents:
(A) activated carbon, albumen, casein, clay, diatomaceous earth, egg-white, isinglass, polyvinylpolypyrrolidone and silicon dioxide,
(B) acacia gum, agar, gelatin and potassium ferrocyanide, at a maximum level of use consistent with good manufacturing practice,
(C) tannic acid at a maximum level of use of 200 parts per million, and
(D) polyvinylpyrrolidone in an amount that does not exceed 2 parts per million in the finished product,
(xiii) caramel at a maximum level of use consistent with good manufacturing practice,
(xiv) brandy, fruit spirit or alcohol derived from the alcoholic fermentation of a food source distilled to not less than 94 per cent alcohol by volume,
(xv) any of the following substances:
(A) carbon dioxide and ozone at a maximum level of use consistent with good manufacturing practice, and
(B) oxygen,
(xvi) sorbic acid or salts thereof, not exceeding 500 parts per million calculated as sorbic acid,
(xvii) malolactic bacteria from the genera Lactobacillus, Leuconostoc and Pediococcus,
(xviii) copper sulphate in such a quantity that the content of copper in the finished product shall not exceed 0.0001 per cent,
(xix) nitrogen, and
(xx) oak chips and particles; and
(c) prior to final filtration may be treated with
(i) a strongly acid cation exchange resin in the sodium ion form, or
(ii) a weakly basic anion exchange resin in the hydroxyl ion form.
- SOR/78-402, s. 1
- SOR/81-565, s. 1
- SOR/84-300, ss. 14(F), 15(E)
- SOR/2006-91, s. 1
- SOR/2008-142, s. 1(F)
- SOR/2010-143, s. 39(E)
- SOR/2016-74, s. 5(E)
B.02.101 No person shall sell wine that contains more than 0.24 per cent weight by volume of volatile acidity calculated as acetic acid, as determined by official method FO-2, Determination of Volatile Acidity of Wine, Cider and Champagne Cider, October 15, 1981.
- SOR/82-768, s. 2
- SOR/2006-91, s. 2
B.02.102 [S]. Fruit spirit shall be an alcoholic distillate obtained from wine, fruit wine, grape pomace or fruit pomace.
B.02.103 [S]. Fruit Wine, or (naming the fruit) Wine shall be the product of the alcoholic fermentation of the juice of sound ripe fruit other than grape, and in all other respects shall meet the requirements of the standard for wine as prescribed by section B.02.100.
B.02.104 [S]. Vermouth shall be wine to which has been added bitters, aromatics or other botanical substances or a flavouring preparation, and shall contain not more than 20 per cent absolute alcohol by volume.
- SOR/93-145, s. 17(F)
B.02.105 [S]. Flavoured Wine, Wine Cocktail, Aperitif Wine shall be wine to which has been added herbs, spices, other botanical substances, fruit juices or a flavouring preparation, and shall contain not more than 20 per cent absolute alcohol by volume.
B.02.105A [S]. Flavoured (naming the fruit) Wine, (naming the fruit) Wine Cocktail, or Aperitif (naming the fruit) Wine shall be fruit wine, a mixture of fruit wines, or a mixture of fruit wine and wine to which has been added herbs, spices, other botanical substances, fruit juices or a flavouring preparation, and shall contain not more than 20 per cent absolute alcohol by volume.
B.02.106 [S]. Honey Wine
(a) shall be the product of the alcoholic fermentation of an aqueous solution of honey; and
(b) may have added to it during the course of manufacture any of the following substances:
(i) yeast;
(ii) yeast foods;
(iii) sulphurous acid, including salts thereof, in such quantity that its content in the finished wine shall not exceed
(A) 70 p.p.m. in the free state, or
(B) 350 p.p.m. in the combined state, calculated as sulphur dioxide;
(iv) tartaric or citric acid;
(v) potassium acid tartrate;
(vi) natural botanical flavours;
(vii) fruit spirit or alcohol derived from the alcoholic fermentation of a food source distilled to not less than 94 per cent alcohol by volume;
(viii) caramel;
(ix) carbon dioxide;
(x) activated carbon, clay or tannic acid as fining agents; or
(xi) sorbic acid, and any salts thereof, calculated as sorbic acid, in a quantity such that the content of sorbic acid and its salts in the finished wine does not exceed 500 parts per million.
- SOR/96-241, s. 1
- SOR/2010-94, s. 9(E)
B.02.107 [S]. May Wine shall be wine to which has been added artificial woodruff flavouring preparation.
B.02.108 A clear indication of the country of origin shall be shown on the principal display panel of a wine.
- SOR/84-300, s. 16(E)
Cider
B.02.120 [S]. Cider
(a) shall
(i) be the product of the alcoholic fermentation of apple juice, and
(ii) contain not less than 2.5 per cent and not more than 13.0 per cent absolute alcohol by volume; and
(b) may have added to it during the course of manufacture
(i) yeast,
(ii) concentrated apple juice,
(iii) sugar, dextrose, invert sugar, glucose, glucose solids, or aqueous solutions thereof,
(iv) yeast foods,
(v) sulphurous acid, including salts thereof, in such quantity that its content in the finished cider shall not exceed
(A) 70 parts per million in the free state, or
(B) 350 parts per million in the combined state, calculated as sulphur dioxide,
(vi) tartaric acid and potassium tartrate,
(vii) citric acid,
(viii) lactic acid,
(ix) pectinase and amylase,
(x) ascorbic or erythorbic acid, or salts thereof,
(xi) any of the following fining agents:
(A) activated carbon,
(B) clay,
(C) diatomaceous earth,
(D) gelatin,
(E) albumen,
(F) sodium chloride,
(G) silica gel,
(H) casein,
(I) tannic acid not exceeding 200 parts per million, or
(J) polyvinylpyrrolidone not exceeding two parts per million in the finished product,
(xii) caramel,
(xiii) brandy, fruit spirit or alcohol derived from the alcoholic fermentation of a food source distilled to not less than 94 per cent alcohol by volume,
(xiv) carbon dioxide,
(xv) oxygen,
(xvi) ozone, or
(xvii) sorbic acid or salts thereof, not exceeding 500 parts per million, calculated as sorbic acid.
- SOR/81-565, s. 2
- SOR/84-300, s. 17(E)
B.02.122 [S]. Champagne Cider shall be cider that is impregnated with carbon dioxide under pressure by
(a) conducting the afterpart of the fermentation in closed vessels, or
(b) secondary fermentation in closed vessels with or without the addition of sugar, dextrose, invert sugar, glucose or glucose solids or aqueous solutions thereof,
and shall contain not less than seven per cent absolute alcohol by volume.
- SOR/84-300, s. 18
B.02.123 No person shall sell cider or champagne cider that has more than 0.2 per cent weight by volume of volatile acidity calculated as acetic acid, as determined by official method FO-2, Determination of Volatile Acidity of Wine, Cider and Champagne Cider, October 15, 1981.
- SOR/82-768, s. 3
Beer
B.02.130 [S]. (1) Beer
(a) shall be the product of the alcoholic fermentation by yeast, or a mixture of yeast and other micro-organisms, an infusion of barley or wheat malt and hops or hop extract in potable water;
(b) shall contain at most 4% of residual sugars; and
(c) may have added to it during the course of manufacture any of the following ingredients:
(i) cereal grain,
(ii) honey, maple syrup, fruit, fruit juice or any other source of carbohydrates,
(iii) herbs and spices,
(iv) salt,
(v) flavouring preparations,
(vi) pre-isomerized hop extract,
(vii) reduced isomerized hop extract, and
(viii) food additives to which a marketing authorization applies and that are set out in the Lists of Permitted Food Additives published on the Health Canada website.
(2) The name of any flavouring preparation added to a beer shall form part of the common name of the beer.
- SOR/88-418, s. 2
- SOR/92-92, s. 1
- SOR/96-483, s. 1
- SOR/2006-91, s. 3
- SOR/2019-98, s. 3
B.02.131 [Repealed, SOR/2019-98, s. 4]
B.02.132 The qualified common name or common name set out in column II of the table to this section shall be used in any advertisement and on the label of a beer that contains the percentage of alcohol by volume set out in column I.
TABLE
| Column I | Column II | |
|---|---|---|
| Item | Percentage Alcohol by Volume | Qualified Common Name or Common Name |
| 1 | 1.1 to 2.5 | Extra Light Beer |
| 2 | 2.6 to 4.0 | Light Beer |
| 3 | 4.1 to 5.5 | Beer |
| 4 | 5.6 to 8.5 | Strong Beer |
| 5 | 8.6 or more | Extra Strong Beer |
- SOR/88-418, s. 2
- SOR/2019-98, s. 5
B.02.133 [S]. In this Division, hop extract means an extract derived from hops by a process employing the solvent
(a) hexane, methanol, or methylene chloride in such a manner that the hop extract does not contain more than 2.2 per cent of the solvent used; or
(b) carbon dioxide or ethyl alcohol in an amount consistent with good manufacturing practice.
- SOR/86-89, s. 1
- SOR/88-418, s. 3
B.02.134 [S]. (1) In this Division, pre-isomerized hop extract means an extract derived from hops by
(a) the use of one of the following solvents:
(i) hexane,
(ii) carbon dioxide, or
(iii) ethanol; and
(b) the subsequent isolation of the alpha acids and their conversion to isomerized alpha acids by means of diluted alkali and heat.
(2) For the purposes of paragraph (1)(b), the residues of hexane shall not exceed 1.5 parts per million per per cent iso-alpha acid content of the pre-isomerized hop extract.
- SOR/88-418, s. 4
B.02.135 [S]. In this Division, reduced isomerized hop extract means
(a) tetrahydroisohumulones derived from hops
(i) by isomerization and reduction of humulones (alpha-acids) by means of hydrogen and a catalyst, or
(ii) by reduction of lupulones (beta-acids) by means of hydrogen and a catalyst, followed by oxidation and isomerization;
(b) hexahydroisohumulones derived from hops by reduction of tetrahydroisohumulones by means of sodium borohydride; and
(c) dihydroisohumulones derived from hops by reduction of isoalpha acids by means of sodium borohydride.
- SOR/96-483, s. 2
- SOR/2000-352, s. 1
DIVISION 3Baking Powder
B.03.001 In this Division, acid-reacting material means one or any combination of
(a) lactic acid or its salts;
(b) tartaric acid or its salts;
(c) acid salts of phosphoric acid; and
(d) acid compounds of aluminum.
B.03.002 [S]. Baking Powder shall be a combination of sodium or potassium bicarbonate, an acid-reacting material, starch or other neutral material, may contain an anticaking agent and shall yield not less than 10 per cent of its weight of carbon dioxide, as determined by official method FO-3, Determination of Carbon Dioxide in Baking Powder, October 15, 1981.
- SOR/82-768, s. 4
- SOR/92-626, s. 12
DIVISION 4Cocoa and Chocolate Products
B.04.001 The definitions in this section apply in this Division.
- chocolate product
chocolate product means a product derived from one or more cocoa products and includes chocolate, bittersweet chocolate, semi-sweet chocolate, dark chocolate, sweet chocolate, milk chocolate and white chocolate. (produit de chocolat)
- cocoa product
cocoa product means a product derived from cocoa beans and includes cocoa nibs, cocoa liquor, cocoa mass, unsweetened chocolate, bitter chocolate, chocolate liquor, cocoa, low fat cocoa, cocoa powder and low fat cocoa powder. (produit du cacao)
- milk ingredient
milk ingredient means one or any combination of
(a) the following products for which a standard is prescribed in this Part, namely,
(i) milk or whole milk,
(ii) skim milk,
(iii) partly skimmed milk or partially skimmed milk,
(iv) sterilized milk,
(v) condensed milk or sweetened condensed milk,
(vi) evaporated milk,
(vii) evaporated skim milk or concentrated skim milk,
(viii) evaporated partly skimmed milk or concentrated partly skimmed milk,
(ix) milk powder or whole milk powder or dry whole milk or powdered whole milk,
(x) skim milk powder or dry skim milk,
(xi) skim milk with added milk solids,
(xii) partly skimmed milk with added milk solids or partially skimmed milk with added milk solids,
(xiii) malted milk or malted milk powder,
(xiv) butter, and
(xv) cream; and
(b) the following products for which a standard is not prescribed by this Part, namely
(i) reconstituted milk or whole milk,
(ii) reconstituted skim milk,
(iii) reconstitued partly skimmed milk,
(iv) partly skimmed milk powder,
(v) buttermilk,
(vi) butter oil, and
(vii) reconstituted cream. (ingrédient laitier)
- sweetening ingredient
sweetening ingredient means any one or any combination of sweetening agents, except for icing sugar. (ingrédient édulcorant)
- SOR/97-263, s. 2
B.04.002 [S]. Cocoa Beans shall be the seeds of Theobroma cacao L. or a closely related species.
- SOR/97-263, s. 2
B.04.003 [S]. Cocoa Nibs shall be the product prepared by removing the shell from cleaned cocoa beans, of which the residual shell content may not exceed 1.75 per cent by mass, calculated to an alkali free basis if the nibs or the cocoa beans from which the nibs were prepared have been processed with alkali, as determined by the method prescribed in the Official Methods of Analysis of the Association of Official Analytical Chemists, 12th Ed. (1975), sections 13.010 to 13.014, under the heading “Shell in Cacao Nibs — Official Final Action”, published by the Association of Official Analytical Chemists, in Washington.
- SOR/97-263, s. 2
B.04.004 [S]. Cocoa Liquor, Cocoa Mass, Unsweetened Chocolate, Bitter Chocolate or Chocolate Liquor shall
(a) be the product obtained from the mechanical disintegration of the cocoa nib with or without removal or addition of any of its constituents; and
(b) contain not less than 50 per cent cocoa butter.
- SOR/97-263, s. 2
B.04.005 (1) Cocoa products may be processed with one or more of the following pH-adjusting or alkalizing agents:
(a) hydroxides of ammonia, carbonates of ammonia, bicarbonates of ammonia, hydroxides of sodium, carbonates of sodium, bicarbonates of sodium, hydroxides of potassium, carbonates of potassium or bicarbonates of potassium;
(b) carbonates of magnesium or hydroxides of magnesium; and
(c) carbonates of calcium.
(2) The quantity of any one pH-adjusting agent referred to in paragraphs (1)(a) to (c) shall not exceed the maximum level of use for that agent set out in column III of an item of Table X to section B.16.100.
(3) The total mass of the pH-adjusting agents referred to in paragraphs (1)(a) to (c) shall not be greater in neutralizing value, calculated from the respective masses of those agents, than the neutralizing value of five parts by mass of anhydrous potassium carbonate for each 100 parts by mass of cocoa product, calculated on a fat-free basis.
(4) Cocoa products may be processed with one or more of the following pH-adjusting or neutralizing agents, added as such or in aqueous solution:
(a) phosphoric acid;
(b) citric acid; and
(c) tartaric acid.
(5) The total mass of pH-adjusting agents referred to in subsection (4) shall not exceed in neutralizing value, calculated from the respective masses of those agents, the appropriate maximum levels of use set out in column III of Table X to section B.16.100.
(6) For the purpose of subsection (5),
(a) the total quantity of phosphoric acid shall not be greater than 0.5 part by mass, expressed as P2O5, for each 100 parts by mass of cocoa product, calculated on a fat-free basis; and
(b) the total quantity of citric acid and tartaric acid, singly or in combination, shall not be greater than 1.0 part by mass for each 100 parts by mass of cocoa product, calculated on a fat-free basis.
- SOR/97-263, s. 2
- SOR/2012-43, s. 1
B.04.006 [S]. Chocolate, Bittersweet Chocolate, Semi-sweet Chocolate or Dark Chocolate
(a) shall be one or more of the following combined with a sweetening ingredient, namely,
(i) cocoa liquor,
(ii) cocoa liquor and cocoa butter, and
(iii) cocoa butter and cocoa powder;
(b) shall contain not less than 35 per cent total cocoa solids, of which
(i) not less than 18 per cent is cocoa butter, and
(ii) not less than 14 per cent is fat-free cocoa solids; and
(c) may contain
(i) less than 5 per cent total milk solids from milk ingredients,
(ii) spices,
(iii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iv) salt, and
(v) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of chocolate product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin,
(C) ammonium salts of phosphorylated glycerides,
(D) polyglycerol esters of interesterified castor oil fatty acids, and
(E) sorbitan monostearate.
- SOR/79-664, s. 2
- SOR/97-263, s. 2
B.04.007 [S]. Sweet Chocolate
(a) shall be one or more of the following combined with a sweetening ingredient, namely,
(i) cocoa liquor,
(ii) cocoa liquor and cocoa butter, and
(iii) cocoa butter and cocoa powder;
(b) shall contain not less than 30 per cent total cocoa solids, of which
(i) 18 per cent is cocoa butter, and
(ii) 12 per cent is fat-free cocoa solids; and
(c) may contain
(i) less than 12 per cent total milk solids from milk ingredients,
(ii) spices,
(iii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iv) salt, and
(v) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of chocolate product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin,
(C) ammonium salts of phosphorylated glycerides,
(D) polyglycerol esters of interesterified castor oil fatty acids, and
(E) sorbitan monostearate.
- SOR/97-263, s. 2
B.04.008 [S]. Milk Chocolate
(a) shall be one or more of the following combined with a sweetening ingredient, namely,
(i) cocoa liquor,
(ii) cocoa liquor and cocoa butter, and
(iii) cocoa butter and cocoa powder;
(b) shall contain not less than
(i) 25 per cent total cocoa solids, of which
(A) not less than 15 per cent is cocoa butter, and
(B) not less than 2.5 per cent is fat-free cocoa solids,
(ii) 12 per cent total milk solids from milk ingredients, and
(iii) 3.39 per cent milk fat; and
(c) may contain
(i) less than 5 per cent whey or whey products,
(ii) spices,
(iii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iv) salt, and
(v) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of chocolate product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin,
(C) ammonium salts of phosphorylated glycerides,
(D) polyglycerol esters of interesterified castor oil fatty acids, and
(E) sorbitan monostearate.
- SOR/97-263, s. 2
B.04.009 [S]. White Chocolate
(a) shall contain the following combined together, namely,
(i) not less than 20 per cent cocoa butter,
(ii) not less than 14 per cent total milk solids from milk ingredients, and
(iii) not less than 3.5 per cent milk fat; and
(b) may contain
(i) less than 5 per cent whey or whey products,
(ii) spices,
(iii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iv) salt, and
(v) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of chocolate product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin,
(C) ammonium salts of phosphorylated glycerides,
(D) polyglycerol esters of interesterified castor oil fatty acids, and
(E) sorbitan monostearate.
- SOR/97-263, s. 2
B.04.010 [S]. Cocoa or Cocoa Powder
(a) shall be the product that
(i) is obtained by pulverising the remaining material from partially defatted cocoa liquor by mechanical means, and
(ii) contains not less than 10 per cent cocoa butter; and
(b) may contain
(i) spices,
(ii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iii) salt, and
(iv) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of cocoa product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin, and
(C) ammonium salts of phosphorylated glycerides.
- SOR/82-768, s. 5
- SOR/97-263, s. 2
B.04.011 [S]. Low Fat Cocoa or Low Fat Cocoa Powder
(a) shall be the product that:
(i) is obtained by pulverising the remaining material from partially defatted cocoa liquor by mechanical means, and
(ii) contains less than 10 per cent cocoa butter; and
(b) may contain
(i) spices,
(ii) flavouring preparations, other than those that imitate the flavour of chocolate or milk, to balance flavour,
(iii) salt, and
(iv) any of the following emulsifying agents, which singly shall not exceed the maximum level of use set out in column III of Table IV to section B.16.100, and in combination shall not exceed 1.5 per cent by mass of cocoa product, namely,
(A) mono-glycerides and mono- and diglycerides,
(B) lecithin and hydroxylated lecithin, and
(C) ammonium salts of phosphorylated glycerides.
- SOR/82-768, s. 6
- SOR/97-263, s. 2
B.04.012 No person shall sell a cocoa product or a chocolate product unless it is free from bacteria of the genus Salmonella as determined by official method MFO-11, Microbiological Examination of Cocoa and Chocolate, November 30, 1981.
- SOR/97-263, s. 2
DIVISION 5Coffee
B.05.001 [S]. Green Coffee, Raw Coffee or Unroasted Coffee shall be the seed of Coffee arabica L., C. liberica Hiern, or C. robusta Chev., freed from all but a small portion of its spermoderm.
B.05.002 [S]. Roasted Coffee or Coffee shall be roasted green coffee, and shall contain not less than 10 per cent fat, and may contain not more than six per cent total ash.
B.05.003 [S]. Decaffeinated (indicating the type of coffee)
(a) shall be coffee of the type indicated, from which caffeine has been removed and that, as a result of the removal, contains not more than
(i) 0.1 per cent caffeine, in the case of decaffeinated raw coffee and decaffeinated coffee, or
(ii) 0.3 per cent caffeine, in the case of decaffeinated instant coffee; and
(b) may have been decaffeinated by means of extraction solvents set out in Table XV to Division 16.
- SOR/90-443, s. 1
DIVISION 6
Food Colours
B.06.001 In this Division,
- diluent
diluent means any substance other than a synthetic colour present in a colour mixture or preparation; (diluant)
- dye
dye means the principal dye and associated subsidiary and isomeric dyes contained in a synthetic colour; (pigment)
- mixture
mixture means a mixture of two or more synthetic colours or a mixture of one or more synthetic colours with one or more diluents; (mélange)
- official method FO-7
official method FO-7[Repealed, SOR/2016-305, s. 49]
- official method FO-8
official method FO-8[Repealed, SOR/2016-305, s. 49]
- official method FO-9
official method FO-9[Repealed, SOR/2016-305, s. 49]
- official method FO-10
official method FO-10[Repealed, SOR/2016-305, s. 49]
- official method FO-11
official method FO-11[Repealed, SOR/2016-305, s. 49]
- official method FO-12
official method FO-12[Repealed, SOR/2016-305, s. 49]
- official method FO-13
official method FO-13[Repealed, SOR/2016-305, s. 49]
- official method FO-14
official method FO-14[Repealed, SOR/2016-305, s. 49]
- official method FO-15
official method FO-15[Repealed, SOR/2016-305, s. 49]
- preparation
preparation means a preparation of one or more synthetic colours containing less than three per cent dye and sold for household use; (préparation)
- synthetic colour
synthetic colour means any organic food colour, other than caramel, that is produced by chemical synthesis and that has no counterpart in nature. (colorant synthétique)
- SOR/80-500, s. 2
- SOR/84-440, s. 1
- SOR/2016-305, s. 49
B.06.002 No person shall sell a food, other than a synthetic colour, mixture, preparation or flavouring preparation, that contains, when prepared for consumption according to label directions, more than
(a) 300 parts per million of Allura Red, Amaranth, Erythrosine, Indigotine, Sunset Yellow FCF or Tartrazine or any combination of those colours unless a higher maximum level of use is specified in column III of item 3 of Table III to section B.16.100;
(b) 100 parts per million of Fast Green FCF or Brilliant Blue FCF or any combination of those colours;
(c) 300 parts per million of any combination of the synthetic colours named in paragraphs (a) and (b) within the limits set by those paragraphs; or
(d) 150 parts per million of Ponceau SX.
- SOR/80-500, s. 2
- SOR/84-440, s. 2
- SOR/86-178, s. 1(F)
- SOR/2007-75, s. 1
B.06.003 [Repealed, SOR/2016-305, s. 50]
B.06.004 [Repealed, SOR/2016-305, s. 50]
B.06.005 [Repealed, SOR/2016-305, s. 50]
B.06.006 [Repealed, SOR/2016-305, s. 50]
B.06.007 No person shall sell a preparation for use in or upon food unless
(a) the label carries the words “Food Colour Preparation” on its principal display panel; and
(b) in the case of a liquid preparation, the container has a capacity of 60 ml or less and will permit dropwise discharge only.
- SOR/80-500, s. 2
B.06.008 [Repealed, SOR/2016-305, s. 51]
B.06.009 to B.06.013 [Repealed, SOR/80-500, s. 2]
B.06.021 [Repealed, SOR/2016-305, s. 52]
B.06.022 [Repealed, SOR/2016-305, s. 52]
B.06.023 [Repealed, SOR/2016-305, s. 52]
B.06.024 [Repealed, SOR/2016-305, s. 52]
B.06.025 [Repealed, SOR/2016-305, s. 52]
B.06.031 [Repealed, SOR/2016-305, s. 52]
B.06.032 [Repealed, SOR/2016-305, s. 52]
B.06.033 [Repealed, SOR/2016-305, s. 52]
B.06.041 [Repealed, SOR/2016-305, s. 53]
B.06.042 [Repealed, SOR/2016-305, s. 53]
B.06.043 [S]. Ponceau SX shall be the disodium salt of 2-(5-sulpho-2,4-xylylazo)-1-naphthol-4-sulphonic acid, shall contain not less than 85% dye and may contain not more than
(a) 0.2% water insoluble matter;
(b) 0.2% combined ether extracts;
(c) 1.0% subsidiary dyes;
(d) 0.5% intermediates;
(e) 3 parts per million of arsenic; and
(f) 10 parts per million of lead.
- SOR/82-768, s. 9
- SOR/84-440, s. 3
- SOR/2016-305, s. 54
B.06.044 [Repealed, SOR/2016-305, s. 54]
B.06.045 [Repealed, SOR/2016-305, s. 54]
B.06.046 [Repealed, SOR/2016-305, s. 54]
B.06.049 [Repealed, SOR/2016-305, s. 54]
B.06.050 [Repealed, SOR/2016-305, s. 54]
B.06.051 [Repealed, SOR/2016-305, s. 54]
B.06.053 [S]. Citrus Red No. 2 shall be 1-(2,5-dimethoxyphenylazo)-2-naphthol, shall contain not less than 98% dye and may contain not more than
(a) 0.5% volatile matter (at 100°C);
(b) 0.3% sulphated ash;
(c) 0.3% water soluble matter;
(d) 0.5% carbon tetrachloride insoluble matter;
(e) 0.05% uncombined intermediates;
(f) 2.0% subsidiary dyes;
(g) 1 part per million of arsenic; and
(h) 10 parts per million of lead.
- SOR/82-768, s. 12
- SOR/84-440, s. 3
- SOR/2016-305, s. 55
B.06.061 The lake of any water soluble synthetic colour that is referred to in section 2 of the Marketing Authorization for Food Additives That May Be Used as Colouring Agents shall be the calcium or aluminum salt of the respective colour extended on alumina.
- SOR/82-1071, s. 3
- SOR/84-300, s. 20
- SOR/87-640, s. 2
- SOR/2016-305, s. 56
B.06.062 [Repealed, SOR/82-768, s. 13]
DIVISION 7Spices, Dressings and Seasonings
B.07.001 [S]. Allspice or Pimento, whole or ground, shall be the dried, full but unripe whole berries of Pimenta dioica (L) Merr. and shall contain
(a) not more than
(i) 25 per cent crude fibre,
(ii) 5.5 per cent total ash,
(iii) 0.4 per cent ash insoluble in hydrochloric acid, and
(iv) 12 per cent moisture; and
(b) not less than 2.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.002 [S]. Anise or Anise Seed, whole or ground, shall be the dried fruit of Pimpinella anisum L. and shall contain
(a) not more than
(i) nine per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than two millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.003 [S]. Basil or Sweet Basil, whole or ground, shall be the dried leaves of Ocimum basilicum L. and shall contain
(a) not more than
(i) 15 per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than 0.2 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.004 [S]. Bay Leaves or Laurel Leaves, whole or ground, shall be the dried leaves of Laurus nobilis L. and shall contain
(a) not more than
(i) 4.5 per cent total ash,
(ii) 0.5 per cent ash insoluble in hydrochloric acid, and
(iii) seven per cent moisture; and
(b) not less than one millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.005 [S]. Caraway or Caraway Seed, whole or ground, shall be the dried fruit of the caraway plant, Carum carvi L. and shall contain
(a) not more than
(i) eight per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 11.5 per cent moisture; and
(b) not less than two millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.006 [S]. Cardamom or Cardamom Seed, bleached or green, whole or ground, shall be the dried ripe fruit of Elettaria cardamomum Maton and shall contain
(a) not more than
(i) eight per cent total ash,
(ii) three per cent ash insoluble in hydrochloric acid, and
(iii) 13 per cent moisture; and
(b) not less than three millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.007 [S]. Cayenne Pepper or Cayenne, whole or ground,
(a) shall be the dried ripe fruit of Capsicum frutescens L., Capsicum baccatum L., or other small-fruited species of Capsicum and shall contain not more than
(i) 1.5 per cent starch,
(ii) 28 per cent crude fibre,
(iii) 10 per cent total ash,
(iv) 1.5 per cent ash insoluble in hydrochloric acid, and
(v) 10 per cent moisture; and
(b) may contain silicon dioxide as an anti-caking agent in an amount not exceeding 2.0 per cent.
- SOR/79-659, s. 1
- SOR/84-17, s. 2
B.07.008 [S]. Celery Salt
(a) shall be a combination of
(i) ground celery seed or ground dehydrated celery, and
(ii) salt in an amount not exceeding 75 per cent; and
(b) may contain silicon dioxide as an anticaking agent in an amount not exceeding 0.5 per cent.
- SOR/79-659, s. 1
B.07.009 [S]. Celery Seed, whole or ground, shall be the dried ripe fruit of Apium graveolens L. and shall contain
(a) not more than
(i) 12 per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than 1.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.010 [S]. Celery Pepper
(a) shall be a combination of
(i) ground celery seed or ground dehydrated celery, and
(ii) ground black pepper in an amount not exceeding 70 per cent; and
(b) may contain silicon dioxide as an anticaking agent in an amount not exceeding 0.5 per cent.
- SOR/79-659, s. 1
B.07.011 [S]. Cinnamon or Cassia, whole or ground, shall be the dried bark of trees of the genus Cinnamomum of Species C. burmanni Blume, C. loureirii Nees or C. Cassia Blume and shall contain
(a) not more than
(i) six per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) 13 per cent moisture; and
(b) not less than 1.2 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.012 [S]. Ceylon Cinnamon, whole or ground, shall be cinnamon obtained exclusively from Cinnamomum zeylanicum Nees.
- SOR/79-659, s. 1
B.07.013 [S]. Cloves, whole or ground, shall be the dried unopened flower buds of Eugenia caryophyllus (Spreng) and shall contain
(a) not more than
(i) five per cent clove stems,
(ii) six per cent total ash,
(iii) 0.5 per cent ash insoluble in hydrochloric acid,
(iv) 10 per cent crude fibre, and
(v) eight per cent moisture; and
(b) not less than 13 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.014 [S]. Coriander or Coriander Seed, whole or ground, shall be the dried fruit of Coriandrum sativum L. and shall contain
(a) not more than
(i) seven per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than 0.3 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.015 [S]. Cumin or Cumin Seed, whole or ground, shall be the dried seeds of Cuminum cyminum L. and shall contain
(a) not more than
(i) 9.5 per cent total ash,
(ii) 1.5 per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than 2.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.016 [S]. Curry Powder shall be any combination of
(a) turmeric with spices and seasoning; and
(b) salt in an amount not exceeding five per cent.
- SOR/79-659, s. 1
B.07.017 [S]. Dill Seed, whole or ground, shall be the dried fruit of Anethum graveolens L., or Anethum sowa D.C. and shall contain
(a) not more than
(i) 10 per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than two millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.018 [S]. Fennel or Fennel Seed, whole or ground, shall be the dried ripe fruit of Foeniculum vulgare Mill. and shall contain
(a) not more than
(i) 10 per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than one millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.019 [S]. Fenugreek, whole or ground, shall be the dried ripe fruit of Trigonella foenumgraecum L. and shall contain not more than
(a) five per cent total ash;
(b) one per cent ash insoluble in hydrochloric acid; and
(c) 10 per cent moisture.
- SOR/79-659, s. 1
B.07.020 [S]. Garlic Salt
(a) shall be a combination of
(i) powdered dehydrated garlic, and
(ii) salt in an amount not exceeding 75 per cent; and
(b) may contain one or more of the following anticaking agents in a total amount not exceeding two per cent:
calcium aluminum silicate, calcium phosphate tribasic, calcium silicate, calcium stearate, magnesium carbonate, magnesium silicate, magnesium stearate, silicone dioxide in an amount not exceeding one per cent and sodium aluminum silicate.
- SOR/79-659, s. 1
B.07.021 [S]. Ginger whole or ground, shall be the washed and dried or decorticated rhizome of Zingiber officinale Roscoe and shall contain
(a) not more than
(i) seven per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 12.5 per cent moisture; and
(b) not less than 1.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.022 [S]. Mace, whole or ground, shall be the dried arillus of Myristica fragrans Houttyn and shall contain
(a) not more than
(i) 3.5 per cent total ash,
(ii) 0.5 per cent ash insoluble in hydrochloric acid, and
(iii) eight per cent moisture, and
(b) not less than 11 millilitres volatile oil per 100 grams of spice,
and the total non-volatile extracts obtainable therefrom by
(c) ethyl ether, shall not be less than 20 per cent nor more than 35 per cent, and
(d) ethyl ether, after preliminary extraction with petroleum ether, shall not exceed five per cent.
- SOR/79-659, s. 1
B.07.023 [S]. Marjoram, whole or ground, shall be the dried leaves, with or without a small proportion of the flowering tops, of Marjorana hortensis Moench and shall contain
(a) not more than
(i) 10 per cent stems and foreign material of plant origin,
(ii) 13.5 per cent total ash,
(iii) 4.5 per cent ash insoluble in hydrochloric acid, and
(iv) 10 per cent moisture; and
(b) not less than 0.7 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.024 [S]. Mustard Seed shall be the seed of Sinapis alba, Brassica hirta Moench, Brassica juncea (L) Cosson or Brassica nigra and shall contain
(a) not more than
(i) seven per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 11 per cent moisture; and
(b) not less than 25 per cent non-volatile ether extract.
- SOR/79-659, s. 1
B.07.025 [S]. Mustard, Mustard Flour or Ground Mustard shall be powdered mustard seed
(a) made from mustard seed from which
(i) most of the hulls have been removed, and
(ii) a portion of the fixed oil may have been removed; and
(b) that contains not more than
(i) 1.5 per cent starch, and
(ii) eight per cent total ash, on an oil-free basis.
- SOR/79-659, s. 1
- SOR/2010-142, s. 3(F)
B.07.026 [S]. Nutmeg, whole or ground, shall be the dried seed of Myristica fragrans Houttyn and may, prior to grinding, have a thin coating of lime and shall contain
(a) not more than
(i) three per cent total ash,
(ii) 0.5 per cent ash insoluble in hydrochloric acid, and
(iii) eight per cent moisture; and
(b) not less than
(i) 25 per cent non-volatile ether extract, and
(ii) 6.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.027 [S]. Onion Salt
(a) shall be a combination of
(i) powdered dehydrated onion, and
(ii) salt in an amount not exceeding 75 per cent; and
(b) may contain one or more of the following anticaking agents in a total amount not exceeding two per cent:
calcium aluminum silicate, calcium phosphate tribasic, calcium silicate, calcium stearate, magnesium carbonate, magnesium silicate, magnesium stearate, silicon dioxide in an amount not exceeding one per cent and sodium aluminum silicate.
- SOR/79-659, s. 1
B.07.028 [S]. Oregano, whole or ground, shall be the dried leaves of Origanum vulgar L. or Origanum Spp. and shall contain
(a) not more than
(i) 10 per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than 2.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.029 [S]. Paprika shall be the dried, ground ripe fruit of Capsicum annuum L. and
(a) shall contain not more than
(i) 23 per cent crude fibre,
(ii) 8.5 per cent total ash,
(iii) one per cent ash insoluble in hydrochloric acid, and
(iv) 12 per cent moisture; and
(b) may contain silicon dioxide as an anti-caking agent in an amount not exceeding 2.0 per cent.
- SOR/79-659, s. 1
- SOR/84-17, s. 3
B.07.030 [S]. Black Pepper or Peppercorn, whole or ground, shall be the dried, immature berry of Piper nigrum L. and shall contain
(a) not more than
(i) seven per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) 12 per cent moisture;
(b) not less than
(i) six per cent non-volatile methylene chloride extract,
(ii) 30 per cent pepper starch, and
(iii) 2.0 millilitres volatile oil per 100 grams of spice; and
(c) when ground, all the parts of the berry in their normal proportions.
- SOR/79-659, s. 1
B.07.031 [S]. White Pepper, whole or ground, shall be the dried, mature berry of Piper nigrum L., from which the outer coating is and the inner coating may be removed and shall contain
(a) not more than
(i) five per cent crude fibre,
(ii) 2.5 per cent total ash,
(iii) 0.3 per cent ash insoluble in hydrochloric acid, and
(iv) 15 per cent moisture; and
(b) not less than
(i) six per cent non-volatile methylene chloride extract,
(ii) 52 per cent pepper starch, and
(iii) one millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.032 [S]. Poppy seed shall be the dried seed of Papaver somniferum L. and shall contain
(a) not more than
(i) seven per cent total ash, and
(ii) one per cent ash insoluble in hydrochloric acid; and
(b) not less than 40 per cent non-volatile ether extract.
- SOR/79-659, s. 1
B.07.033 [S]. Rosemary, whole or ground, shall be the dried leaves of Rosemarinus officinalis L. and shall contain
(a) not more than
(i) 7.5 per cent total ash,
(ii) one per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than 1.2 millilitres volatile oil per 100 grams of spice.
- SOR/78-637, s. 1
- SOR/79-659, s. 1
B.07.034 [S]. Sage, whole or ground, shall be the dried leaves of the sage plant Salvia officinalis L., Salvia triloba L. or Salvia lavandulaefolia Vahl. and shall contain
(a) not more than
(i) 12 per cent stems excluding petioles and foreign material of plant origin,
(ii) 10 per cent total ash,
(iii) one per cent ash insoluble in hydrochloric acid, and
(iv) 10 per cent moisture; and
(b) not less than one millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.035 [S]. Savory, whole or ground, shall be the dried leaves and flowering tops of Satureja hortensis L. or Satureja montana L. and shall contain
(a) not more than
(i) 11 per cent total ash,
(ii) two per cent ash insoluble in hydrochloric acid, and
(iii) 14 per cent moisture; and
(b) not less than 0.8 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.036 [S]. Sesame Seed shall be the dried hulled seed of Sesamum indicum L. and shall contain
(a) not more than eight per cent moisture; and
(b) not less than 50 per cent non-volatile ether extract.
- SOR/79-659, s. 1
- SOR/2010-142, s. 4(F)
B.07.037 [S]. Tarragon, whole or ground, shall be the dried leaves and flowering tops of Artemisia dracunculus L. and shall contain
(a) not more than
(i) 15 per cent total ash,
(ii) 1.5 per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than 0.3 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.038 [S]. Thyme, whole or ground, shall be the dried leaves and flowering tops of the thyme plant Thymus vulgaris L. or Thymus zygis L. and shall contain
(a) not more than
(i) 12 per cent total ash,
(ii) five per cent ash insoluble in hydrochloric acid, and
(iii) nine per cent moisture; and
(b) not less than 0.9 millilitre volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.039 [S]. Turmeric, whole or ground, shall be the dried rhizome of Curcuma longa L. and shall contain
(a) not more than
(i) seven per cent total ash,
(ii) 1.5 per cent ash insoluble in hydrochloric acid, and
(iii) 10 per cent moisture; and
(b) not less than 3.5 millilitres volatile oil per 100 grams of spice.
- SOR/79-659, s. 1
B.07.040 [S]. Mayonnaise, Mayonnaise Dressing or Mayonnaise Salad Dressing
(a) shall be a combination of
(i) vegetable oil,
(ii) whole egg or egg yolk, in liquid, frozen or dried form, and
(iii) vinegar or lemon juice;
(b) may contain
(i) water,
(ii) salt,
(iii) a sweetening agent,
(iv) spice or other seasoning except turmeric or saffron,
(v) citric, tartaric or lactic acid, and
(vi) a sequestering agent; and
(c) shall contain not less than 65 per cent vegetable oil.
- SOR/79-659, s. 1
B.07.041 [S]. French Dressing
(a) shall be a combination of
(i) vegetable oil, and
(ii) vinegar or lemon juice;
(b) may contain
(i) water,
(ii) salt,
(iii) a sweetening agent,
(iv) spice, tomato or other seasoning,
(v) an emulsifying agent,
(vi) whole egg or egg yolk, in liquid, frozen or dried form,
(vii) citric, tartaric or lactic acid, and
(viii) a sequestering agent; and
(c) shall contain not less than 35 per cent vegetable oil.
- SOR/79-659, s. 1
B.07.042 [S]. Salad Dressing
(a) shall be a combination of
(i) vegetable oil,
(ii) whole egg or egg yolk, in liquid, frozen or dried form,
(iii) vinegar or lemon juice, and
(iv) starch, flour, rye flour or tapioca flour or any combination thereof;
(b) may contain
(i) water,
(ii) salt,
(iii) a sweetening agent,
(iv) spice or other seasoning,
(v) an emulsifying agent,
(vi) citric, tartaric or lactic acid, and
(vii) a sequestering agent; and
(c) shall contain not less than 35 per cent vegetable oil.
- SOR/79-659, s. 1
B.07.043 No person shall sell a dressing that contains more than five per cent C22 Monoenoic Fatty Acids calculated as a proportion of the total fatty acids contained in the dressing.
- SOR/79-659, s. 1
DIVISION 8Dairy Products
B.08.001 The foods referred to in this Division are dairy products.
B.08.001.1 The following definitions apply in this Division.
- milk product
milk product means
(a) with respect to butter or whey butter, any of the following products, namely,
(i) partly skimmed milk, skim milk, cream, buttermilk and whey cream, and
(ii) milk in concentrated, dried or reconstituted form and any product referred to in subparagraph (i) in concentrated, dried or reconstituted form;
(b) with respect to cheese, any of the following products, namely,
(i) partly skimmed milk, skim milk, cream, buttermilk, whey and whey cream,
(ii) milk in concentrated, dried, frozen or reconstituted form and any product referred to in subparagraph (i) in concentrated, dried, frozen or reconstituted form,
(iii) butter, butter oil and whey butter,
(iv) any constituent of milk — other than water — singly or in combination with other constituents of milk, and
(v) whey protein concentrate;
(c) with respect to cold-pack cheese food, cold-pack cheese food with (naming the added ingredients), cream cheese spread, cream cheese spread with (naming the added ingredients), processed cheese food, processed cheese food with (naming the added ingredients), processed cheese spread or processed cheese spread with (naming the added ingredients), any of the following products, namely,
(i) butter, whey butter and whey,
(ii) whey protein concentrate, and
(iii) any product referred to in subparagraph (i) in concentrated or dried form; and
(d) with respect to ice milk mix, ice cream mix or sherbet, any of the products referred to in subparagraph (a)(i) or (ii) or (c)(i), (ii) or (iii). (produit du lait)
- ultrafiltered
ultrafiltered, in relation to milk, partly skimmed milk or skim milk, means that the milk, partly skimmed milk or skim milk has been subjected to a process in which it is passed over one or more semi-permeable membranes to partially remove water, lactose, minerals and water-soluble vitamins without altering the whey protein to casein ratio and that results in a liquid product. (ultrafiltré)
- SOR/92-400, s. 1. SOR/97-543, s. 1(F)
- SOR/98-580, s. 1(F)
- SOR/2007-302, s. 1
- SOR/2010-94, s. 2
- SOR/2011-205, s. 41
B.08.002 Except as provided in these Regulations, a dairy product that contains a fat other than milk fat is adulterated.
B.08.002.1 Sections B.08.003 to B.08.028 do not apply to a lacteal secretion obtained from the mammary gland of any animal other than a cow, genus Bos, or a product or derivative of such secretion.
- SOR/85-623, s. 1
B.08.002.2 (1) Subject to subsection (2), no person shall sell the normal lacteal secretion obtained from the mammary gland of the cow, genus Bos, or of any other animal, or sell a dairy product made with any such secretion, unless the secretion or dairy product has been pasteurized by being held at a temperature and for a period that ensure the reduction of the alkaline phosphatase activity so as to meet the tolerances specified in official method MFO-3, Determination of Phosphatase Activity in Dairy Products, dated November 30, 1981.
(2) Subsection (1) does not apply to
(a) cheese; or
(b) any food that is sold for further manufacturing or processing in order to pasteurize it in the manner described in subsection (1).
- SOR/91-549, s. 1
- SOR/95-499, s. 1
Milk
B.08.003 [S]. Milk or Whole Milk
(a) shall be the normal lacteal secretion obtained from the mammary gland of the cow, genus Bos; and
(b) shall contain 2 μg of vitamin D per 100 mL.
- SOR/95-499, s. 2
- SOR/2022-168, s. 22
B.08.004 [S]. Skim Milk
(a) shall be milk that contains not more than 0.3 per cent milk fat;
(b) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A; and
(c) shall contain 2 μg of vitamin D per 100 mL.
- SOR/78-656, s. 1
- SOR/2022-168, s. 23
B.08.005 [S]. Partly Skimmed Milk or Partially Skimmed Milk
(a) shall be derived from milk that has had its fat content reduced by mechanical separation or adjusted by the addition of cream, milk, partly skimmed milk or skim milk, either singly or in combination;
(b) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A; and
(c) shall contain 2 μg of vitamin D per 100 mL.
- SOR/78-656, s. 2
- SOR/2022-168, s. 24
B.08.006 [S]. Milk Fat or Butter Fat shall be the fat of cow’s milk, and shall have
(a) a specific gravity of not less than 0.905 at a temperature of 40°,
(b) a tocopherol content not greater than 50 micrograms per gram, as determined by official method FO-16, Determination of Tocopherol in Milk Fat or Butter Fat, October 15, 1981,
(c) a Reichert-Meissl number not less than 24, and
(d) a Polenske number not exceeding 10 per cent of the Reichert-Meissl number and in no case shall the Polenske number exceed 3.5, and
where the tocopherol content is greater than 50 micrograms per gram or the Polenske number exceeds 10 per cent of the Reichert-Meissl number, there shall be deemed to have been an addition to the milk fat of fat other than that of cow’s milk.
- SOR/82-768, s. 14
B.08.007 [S]. Sterilized Milk
(a) and (b) [Repealed, SOR/97-148, s. 1]
(c) shall contain not less than
(i) 11.75 per cent milk solids, and
(ii) 3.25 per cent milk fat; and
(d) shall contain 2 μg of vitamin D per 100 mL.
- SOR/97-148, s. 1
- SOR/2022-168, s. 25
B.08.008 The percentage of milk fat contained in
(a) partly skimmed milk or partially skimmed milk,
(b) partly skimmed milk with added milk solids or partially skimmed milk with added milk solids, or
(c) evaporated partly skimmed milk or concentrated partly skimmed milk
shall be shown on the principal display panel followed by the words “milk fat” or the abbreviation “B.F.” or “M.F.”.
B.08.009 [S]. Condensed Milk or Sweetened Condensed Milk shall be milk from which water has been evaporated and to which has been added sugar, dextrose, glucose, glucose solids or lactose, or any combination thereof, may contain added vitamin D and shall contain not less than
(a) 28 per cent milk solids; and
(b) eight per cent milk fat.
B.08.010 [S]. Evaporated Milk
(a) shall be milk from which water has been evaporated;
(b) shall contain not less than
(i) 25.0 per cent milk solids, and
(ii) 7.5 per cent milk fat;
(c) shall, notwithstanding sections D.01.009 to D.01.011, contain added vitamin C in such an amount that a reasonable daily intake of the milk contains not less than 60 milligrams and not more than 75 milligrams of vitamin C;
(d) shall contain 2 μg of vitamin D per 100 mL when reconstituted to its original volume; and
(e) may contain
(i) added disodium phosphate or sodium citrate, or both, and
(ii) an emulsifying agent.
- SOR/78-656, s. 3
- SOR/92-400, s. 2
- SOR/2022-168, s. 26
B.08.011 [S]. Evaporated Skim Milk or Concentrated Skim Milk
(a) shall be milk that has been concentrated to at least one-half of its original volume by the removal of water;
(b) shall contain
(i) not more than 0.3 per cent milk fat, and
(ii) not less than 17.0 per cent milk solids other than fat;
(c) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(d) shall, notwithstanding sections D.01.009 to D.01.011, contain added vitamin C in such an amount that a reasonable daily intake of the milk contains not less than 60 milligrams and not more than 75 milligrams of vitamin C;
(e) shall contain 2 μg of vitamin D per 100 mL when reconstituted to its original volume; and
(f) may contain added disodium phosphate or sodium citrate, or both.
B.08.012 [S]. Evaporated Partly Skimmed Milk or Concentrated Partly Skimmed Milk
(a) shall be milk from which part of the milk fat has been removed;
(b) shall be concentrated to at least one-half its original volume by the removal of water;
(c) shall contain not less than 17.0 per cent milk solids other than fat; and
(d) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(e) shall, notwithstanding sections D.01.009 to D.01.011, contain added vitamin C in such an amount that a reasonable daily intake of the milk contains not less than 60 milligrams and not more than 75 milligrams of vitamin C;
(f) shall contain 2 μg of vitamin D per 100 mL when reconstituted to its original volume; and
(g) may contain
(i) an emulsifying agent, and
(ii) added disodium phosphate or sodium citrate, or both.
B.08.013 [S]. Milk Powder, Whole Milk Powder, Dry Whole Milk, or Powdered Whole Milk
(a) shall be dried milk;
(b) shall contain not less than
(i) 95 per cent milk solids, and
(ii) 26 per cent milk fat;
(c) shall contain 2 μg of vitamin D per 100 mL when reconstituted according to directions for use; and
(d) may contain the emulsifying agent lecithin in an amount not exceeding 0.5 per cent.
- SOR/78-656, s. 4
- SOR/83-932, s. 2
- SOR/2022-168, s. 29
B.08.014 [S]. Skim Milk Powder or Dry Skim Milk
(a) shall be dried skim milk;
(b) shall contain not less than 95 per cent milk solids; and
(c) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(d) shall contain 2 μg of vitamin D per 100 mL when reconstituted according to directions for use; and
(e) may contain an anti-foaming agent.
- SOR/78-656, s. 5
- SOR/80-501, s. 2(F)
- SOR/2022-168, s. 30
B.08.014A No person shall sell milk powder, whole milk powder, dry whole milk, powdered whole milk, skim milk powder or dry skim milk unless it is free from bacteria of the genus Salmonella, as determined by official method MFO-12, Microbiological Examination of Milk Powder, November 30, 1981.
- SOR/78-656, s. 6
- SOR/82-768, s. 15
B.08.015 No person shall sell milk, skim milk, partly skimmed milk, (naming the flavour) milk, (naming the flavour) skim milk, (naming the flavour) partly skimmed milk, skim milk with added milk solids, partly skimmed milk with added milk solids, (naming the flavour) skim milk with added milk solids, (naming the flavour) partly skimmed milk with added milk solids, condensed milk, evaporated milk, evaporated skim milk, evaporated partly skimmed milk, milk powder or skim milk powder, in which the vitamin content has been increased by either irradiation or addition unless
(a) in the case of the addition of vitamin D, the menstruum containing the vitamin D contributes not more than 0.01 per cent fat foreign to milk; and
(b) in cases where the vitamin D content is increased by irradiation, the principal display panel of the label carries the statement “Vitamin D Increased” immediately preceding or following the name of the food, without intervening written, printed or graphic matter.
- SOR/88-336, s. 3
B.08.016 [S]. (naming the flavour) Milk
(a) shall be the product made from
(i) milk, milk powder, skim milk, skim milk powder, partly skimmed milk, evaporated milk, evaporated partly skimmed milk, evaporated skim milk or cream or any combination thereof,
(ii) a flavouring preparation, and
(iii) a sweetening agent;
(b) shall contain not less than three per cent milk fat;
(c) shall contain 2 μg of vitamin D per 100 mL;
(d) may contain salt, food colour, lactase, stabilizing agent and not more than 0.5 per cent starch; and
(e) may contain not more than 50,000 total aerobic bacteria per cubic centimetre, as determined by official method MFO-7, Microbiological Examination of Milk, November 30, 1981.
- SOR/78-656, s. 7
- SOR/82-768, s. 16
- SOR/84-762, s. 1
- SOR/2022-168, s. 31
B.08.017 [S]. (naming the flavour) Skim Milk
(a) shall be the product made from
(i) skim milk, skim milk powder or evaporated skim milk or any combination thereof,
(ii) a flavouring preparation, and
(iii) a sweetening agent;
(b) shall contain not more than 0.3 per cent milk fat;
(c) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(d) shall contain 2 μg of vitamin D per 100 mL; and
(e) may contain salt, food colour, lactase, stabilizing agent and not more than 0.5 per cent starch.
- SOR/78-656, s. 8
- SOR/84-762, s. 2
- SOR/2022-168, s. 32
B.08.018 [S]. (naming the flavour) Partly (Partially) Skimmed Milk
(a) shall be the product made from
(i) milk, milk powder, skim milk, skim milk powder, partly skimmed milk, evaporated milk, evaporated partly skimmed milk, evaporated skim milk or cream or any combination thereof,
(ii) a flavouring preparation, and
(iii) a sweetening agent;
(b) shall contain more than 0.3 per cent and less than 3.0 per cent milk fat;
(c) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(d) shall contain 2 μg of vitamin D per 100 mL;
(e) may contain salt, food colour, lactase, stabilizing agent and not more than 0.5 per cent starch; and
(f) may contain not more than 50,000 total aerobic bacteria per cubic centimetre, as determined by official method MFO-7, Microbiological Examination of Milk, November 30, 1981.
- SOR/78-656, s. 9
- SOR/82-768, s. 17
- SOR/84-762, s. 3
- SOR/2022-168, s. 33
B.08.019 [S]. Skim Milk with Added Milk Solids
(a) shall be skim milk to which has been added skim milk powder or evaporated skim milk or both;
(b) shall contain not less than 10 per cent milk solids;
(c) shall contain not more than 0.3 per cent milk fat;
(d) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the dairy product contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A; and
(e) shall contain 2 μg of vitamin D per 100 mL.
- SOR/78-656, s. 10
- SOR/2022-168, s. 34
B.08.020 [S]. Partly Skimmed Milk with Added Milk Solids or Partially Skimmed Milk with Added Milk Solids
(a) shall be partly skimmed milk to which has been added skim milk powder, milk powder, evaporated milk, evaporated partly skimmed milk or evaporated skim milk or any combination thereof;
(b) shall contain not less than 10 per cent milk solids not including fat; and
(c) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A; and
(d) shall contain 2 μg of vitamin D per 100 mL.
B.08.021 [S]. Malted Milk or Malted Milk Powder
(a) shall be the product made by combining milk with the liquid separated from a mash of ground barley malt and meal;
(b) may have added to it, in such manner as to secure the full enzyme action of the malt extract, salt and sodium bicarbonate or potassium bicarbonate;
(c) may have water removed from it; and
(d) shall contain
(i) not less than 7.5 per cent milk fat, and
(ii) not more than 3.5 per cent moisture.
B.08.022 [S]. (naming the flavour) Malted Milk or (naming the flavour) Malted Milk Powder
(a) shall be malted milk or malted milk powder containing a flavouring preparation; and
(b) may contain lactase.
- SOR/84-762, s. 4
B.08.023 [S]. (naming the flavour) Skim Milk with Added Milk Solids
(a) shall be the product made from
(i) skim milk, skim milk powder, or evaporated skim milk or any combination thereof,
(ii) a flavouring preparation, and
(iii) a sweetening agent;
(b) shall contain not less than 10 per cent milk solids not including fat;
(c) shall contain not more than 0.3 per cent milk fat;
(d) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(e) shall contain 2 μg of vitamin D per 100 mL; and
(f) may contain salt, food colour, lactase, stabilizing agent and not more than 0.5 per cent starch.
- SOR/78-656, s. 11
- SOR/84-762, s. 5
- SOR/2022-168, s. 36
B.08.024 No person shall sell milk for manufacture into dairy products if it contains more than
(a) two million total aerobic bacteria per millilitre, as determined by official method MFO-7, Microbiological Examination of Milk, November 30, 1981; or
(b) two milligrams of sediment per 16 fluid ounces, as determined by official method MFO-8, Determination of Sediment in Milk, November 30, 1981.
- SOR/82-768, s. 18
B.08.025 No manufacturer shall purchase milk for manufacture or manufacture milk into other dairy products if he has reason to believe it does not meet the requirements of section B.08.024.
B.08.026 [S]. (naming the flavour) Partly (Partially) Skimmed Milk with Added Milk Solids
(a) shall be the product made from
(i) milk, milk powder, skim milk, skim milk powder, partly skimmed milk, evaporated milk, evaporated partly skimmed milk, evaporated skim milk or cream or any combination thereof,
(ii) a flavouring preparation, and
(iii) a sweetening agent;
(b) shall contain not less than 10 per cent milk solids not including fat;
(c) shall contain more than 0.3 per cent and less than 3.0 per cent milk fat;
(d) shall, notwithstanding sections D.01.009 and D.01.010, contain added vitamin A in such an amount that a reasonable daily intake of the milk contains not less than 1,200 International Units and not more than 2,500 International Units of vitamin A;
(e) shall contain 2 μg of vitamin D per 100 mL;
(f) may contain salt, food colour, lactase, stabilizing agent and not more than 0.5 per cent starch; and
(g) may contain not more than 50,000 total aerobic bacteria per cubic centimetre, as determined by official method MFO-7, Microbiological Examination of Milk, November 30, 1981.
- SOR/78-656, s. 12
- SOR/82-768, s. 19
- SOR/84-300, s. 21(F)
- SOR/84-762, s. 6
- SOR/2022-168, s. 37
B.08.027 Notwithstanding anything contained in these Regulations, the following dairy products that are used in or sold for the manufacture of other foods are not required to contain added vitamins: milk; partly skimmed milk; partially skimmed milk; skim milk; sterilized milk; evaporated milk; evaporated skim milk; concentrated skim milk; evaporated partly skimmed milk; concentrated partly skimmed milk; milk powder; dry whole milk; powdered whole milk; skim milk powder; dry skim milk; partly skimmed milk powder; partially skimmed milk powder; skim milk with added milk solids; partly skimmed milk with added milk solids; and partially skimmed milk with added milk solids.
- SOR/78-656, s. 13
B.08.028 (1) The percentage of milk fat contained in
(a) (naming the flavour) milk,
(b) (naming the flavour) partly (partially) skimmed milk,
(c) (naming the flavour) partly (partially) skimmed milk with added milk solids,
(d) cream, and
(e) sour cream,
shall be shown on the principal display panel followed by the words “milk fat” or the abbreviation “B.F.” or “M.F.”.
(2) In addition to the statement referred to in subsection (1), a person may, on the label of a food referred to in that subsection, make a declaration of the fat content of the food, expressed in grams per serving of stated size.
- SOR/79-23, s. 10
- SOR/88-559, s. 16
- SOR/2010-94, s. 3(F)
- SOR/2016-305, s. 75(F)
Goat’s Milk
B.08.028.1 A lacteal secretion obtained from themammary gland of any animal other than a cow, genus Bos, and a product or derivative of such secretion shall be labelled so as to identify that animal.
- SOR/85-623, s. 2
B.08.029 (1) No person shall sell goat’s milk or goat’s milk powder to which vitamin D has been added unless 100 mL of that food, when ready-to-serve, contains 2 μg of vitamin D.
(2) No person shall sell skimmed or partly skimmed goat’s milk or skimmed or partly skimmed goat’s milk powder to which vitamin A or D has been added unless 100 mL of that food, when ready-to-serve, contains both vitamin A and D in the following amounts:
(a) not less than 140 I.U. and not more than 300 I.U. of vitamin A; and
(b) 2 μg of vitamin D.
(3) No person shall sell evaporated goat’s milk to which any of the following vitamins have been added unless 100 mL of that food, when reconstituted to its original volume, contains each of the following vitamins in the following amounts:
(a) not less than 7 mg and not more than 9 mg of vitamin C;
(b) 2 μg of vitamin D; and
(c) not less than 10 μg and not more than 20 μg of folic acid.
(4) No person shall sell evaporated partly skimmed goat’s milk or evaporated skimmed goat’s milk to which any of the following vitamins have been added unless 100 mL of that food, when reconstituted to its original volume, contains each of the following vitamins in the following amounts:
(a) not less than 140 I.U. and not more than 300 I.U. of vitamin A;
(b) not less than 7 mg and not more than 9 mg of vitamin C;
(c) 2 μg of vitamin D; and
(d) not less than 10 μg and not more than 20 μg of folic acid.
- SOR/85-623, s. 2
- SOR/2022-168, s. 38
Cheese
B.08.030 (1) In this Division, when used with respect to cheese,
- pasteurized source
pasteurized source means milk, skim milk, cream, reconstituted milk powder, reconstituted skim milk powder or any combination thereof that has been pasteurized by being held at a temperature of not less than 61.6°C for a period of not less than 30 minutes, or for a time and a temperature that is equivalent thereto in phosphatase destruction, as determined by official method MFO-3, Determination of Phosphatase Activity in Dairy Products, November 30, 1981; (matière première pasteurisée)
- pickles and relishes
pickles and relishes means foods that meet the standard prescribed in section B.11.051; (cornichons et achards)
- stored
stored means to have been kept or held at a temperature of 2°C or more for a period of 60 days or more from the date of the beginning of the manufacturing process; (entreposé)
- whole
whole means of the original size and shape as manufactured. (entier)
(2) The word “process” may be used in the place of the word “processed” in the following common names: processed (naming the variety) cheese, processed (naming the variety) cheese with (naming the added ingredients), processed cheese food, processed cheese food with (naming the added ingredients), processed cheese spread, and processed cheese spread with (naming the added ingredients).
- SOR/79-752, s. 2
- SOR/82-768, s. 20
- SOR/92-400, s. 3
B.08.031 A cheese made from milk that is the normal lacteal secretion of the mammary gland of animals other than the cow, genus Bos, shall
(a) conform to all requirements in this Division applicable to the variety; and
(b) be labelled to show the source of the milk on the principal display panel.
- SOR/79-752, s. 2
B.08.032 (1) Each of the following foods for which a standard is prescribed, namely,
(a) (naming the variety) cheese,
(b) cheddar cheese,
(c) cream cheese,
(d) whey cheese,
(e) (naming the variety) whey cheese,
(f) cream cheese with (naming the added ingredients),
(g) cream cheese spread,
(h) cream cheese spread with (naming the added ingredients),
(i) processed (naming the variety) cheese,
(j) processed (naming the variety) cheese with (naming the added ingredients),
(k) processed cheese food,
(l) processed cheese food with (naming the added ingredients),
(m) processed cheese spread,
(n) processed cheese spread with (naming the added ingredients),
(o) cold-pack (naming the variety) cheese,
(p) cold-pack (naming the variety) cheese with (naming the added ingredients),
(q) cold-pack cheese food, and
(r) cold-pack cheese food with (naming the added ingredients),
shall be labelled to show on the principal display panel a statement of the percentage of milk fat in the food followed by the words “milk fat” or the abbreviation “B.F.” or “M.F.”, and the percentage of moisture in the food followed by the word “moisture” or “water”.
(2) Subject to subsection (3), no person shall make any direct or indirect reference on the label of a food referred to in subsection (1) to the milk fat content or moisture content of the food except as required by subsection (1).
(3) In addition to the statements referred to in subsection (1), a person may, on the label of a food referred to in that subsection, make a declaration of the fat content of the food, expressed in grams per serving of a stated size.
- SOR/79-752, s. 2
- SOR/88-559, s. 17
- SOR/94-689, s. 2(E)
- SOR/2010-94, s. 8(E)
- SOR/2016-305, s. 75(F)
B.08.033 (1) [S]. (Naming the variety) Cheese, other than cheddar cheese, cream cheese, whey cheese, cream cheese with (naming the added ingredients), cream cheese spread, cream cheese spread with (naming the added ingredients), processed (naming the variety) cheese, processed (naming the variety) cheese with (naming the added ingredients), processed cheese food, processed cheese food with (naming the added ingredients), processed cheese spread, processed cheese spread with (naming the added ingredients), cold-pack (naming the variety) cheese, cold-pack (naming the variety) cheese with (naming the added ingredients), cold-pack cheese food, cold-pack cheese food with (naming the added ingredients), cottage cheese and creamed cottage cheese,
(a) shall
(i) be the product made by coagulating milk, milk products or a combination thereof with the aid of bacteria to form a curd and forming the curd into a homogeneous mass after draining the whey,
(i.1) except for feta cheese, have a casein content that is derived from milk or from ultrafiltered milk, partly skimmed milk, ultrafiltered partly skimmed milk, skim milk, ultrafiltered skim milk or cream, rather than from other milk products,that is at least the following percentage of the total protein content of the cheese, namely,
(A) 63 per cent in the case of Pizza Mozzarella cheese and Part Skim Pizza Mozzarella cheese,
(B) 83 per cent, in the case of Brick cheese, Canadian Style Brick cheese, Canadian Style Munster cheese, Colby cheese, Farmer’s cheese, Jack cheese, Monterey (Monterey Jack) cheese, Mozzarella (Scamorza) cheese, Part Skim Mozzarella (Part Skim Scamorza) cheese, Part Skim Pizza cheese, Pizza cheese, Skim milk cheese and any other variety of cheese not referred to in clause (A) or (C), and
(C) 95 per cent, in the case of any other variety of cheese named in the table to this section,
(i.2) have a whey protein to casein ratio that does not exceed the whey protein to casein ratio of milk,
(ii) possess the physical, chemical and organoleptic properties typical for the variety,
(iii) where it is a cheese of variety named in the table to this section, contain no more than the maximum percentage of moisture shown in Column II thereof for that variety,
(iv) where it is a cheese of a variety named in Part I of the table to this section, contain no less than the minimum percentage of milk fat shown in Column III for that variety, and
(v) where it is cheese of a variety named in Part II of the table to this section, contain no more than the maximum percentage of milk fat shown in Column III for that variety; and
(b) may contain
(i) salt, seasonings, condiments and spices,
(ii) flavouring preparations other than cheese flavouring,
(iii) micro-organisms to aid further ripening,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin or turmeric,
(B) in an amount not exceeding 35 parts per million, beta-apo-8’-carotenal, ethyl beta-apo-8’-carotenoate or a combination thereof, and
(C) in an amount not exceeding 0.10 parts per million, brilliant blue FCF in feta cheese only,
(v) calcium chloride as a firming agent in an amount not exceeding 0.02 per cent of the milk and milk products used,
(vi) paraffin wax as a coating in an amount consistent with good manufacturing practice,
(vii) where potassium nitrate, sodium nitrate or a combination thereof are used for the purpose and in the manner described in subsection (2), residues of potassium nitrate, sodium nitrate or a combination thereof in an amount not exceeding 50 parts per million,
(viii) wood smoke as a preservative in an amount consistent with good manufacturing practice,
(ix) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid,
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively, or
(D) natamycin applied to the surface of the cheese in an amount that does not exceed 20 parts per million or, if the cheese is grated or shredded, 10 parts per million,
(x) in the case of grated or shredded cheese, calcium silicate, microcrystalline cellulose or cellulose, or a combination of them, as an anticaking agent, the total amount not to exceed 2.0 per cent, and
(xi) carbon dioxide as a pH adjusting agent in milk for cheese production, in an amount consistent with good manufacturing practice.
(1.1) A cheese of a variety set out in column I of Part I of the table to this section may contain more than the maximum percentage of moisture set out in column II and less than the minimum percentage of milk fat set out in column III if
(a) a statement or claim set out in column 4 of any of items 12 to 14, 16, 20, 21 and 45 of the Table of Permitted Nutrient Content Statements and Claims is shown on the label of the product as part of the common name; and
(b) the cheese has the characteristic flavour and texture of the named variety of cheese.
(1.2) The reference to “83 per cent” in clause (1)(a)(i.1)(B) shall be read as “78 per cent”, and the reference to “95 per cent” in clause (1)(a)(i.1)(C) shall be read as “90 per cent”, with respect to the named variety of cheese if
(a) a statement or claim set out in column 4 of any of items 12 to 14, 16, 20, 21 and 45 of the Table of Permitted Nutrient Content Statements and Claims is shown on the label of the product as part of the common name; and
(b) the cheese has the characteristic flavour and texture of the named variety of cheese.
(2) Potassium nitrate, sodium nitrate or a combination thereof may be used as a preservative in cheese providing the following requirements are met:
(a) the amount of the salt or combination of salts does not exceed 200 parts per million of the milk and milk products used to make the cheese;
(b) the cheese in which the preservative is used is
(i) mold ripened cheese packed in a hermetically sealed container, or
(ii) ripened cheese
(A) that contains not more than 68 per cent moisture on a fat free basis, and
(B) during the manufacture of which the lactic acid fermentation and salting was completed more than 12 hours after coagulation of the curd by enzymes; and
(c) the salting is, in the case of the cheese described in subparagraph (b)(ii), applied externally as a dry salt or in the form of a brine.
(3) No person shall use an enzyme other than
(a) aminopeptidase derived from Lactococcus lactis, bovine rennet derived from aqueous extracts from the fourth stomach of adult bovine animals, sheep and goats, chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS105), lipase derived from Animal pancreatic tissue; Aspergillus niger var.; Aspergillus oryzae var.; Edible forestomach tissue of calves, kids or lambs; Rhizopus oryzae var. or from Aspergillus oryzae (MLT-2) (pRML 787) (p3SR2); Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)); Rhizopus niveus, milk coagulating enzyme derived from Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)), from Mucor pusillus Lindt by pure culture fermentation process or from Aspergillus oryzae RET-1 (pBoel777), pepsin derived from glandular layer of porcine stomach, phospholipase derived from Aspergillus oryzae (pPFJo142), protease derived from Micrococcus caseolyticus var. or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in the manufacture of any cheese to which subsection (1) applies;
(b) [Repealed, SOR/2010-143, s. 1]
(c) a milk coagulating enzyme derived from Endothia parasitica and enzymes described in paragraph (a), in the manufacture of Emmentaler (Emmental, Swiss) cheese, Mozzarella (Scamorza) cheese and Part Skim Mozzarella (Part Skim Scamorza) cheese;
(d) a milk coagulating enzyme derived from Endothia parasitica and enzymes described in paragraph (a), in the manufacture of Parmesan cheese and Romano cheese;
(e) protease derived from Aspergillus oryzae var., Aspergillus niger var. or Bacillus subtilis var., in the manufacture of Colby cheese; and
(f) lysozyme derived from egg-white.
(3.1) No person shall use an enzyme referred to in subsection (3) at a level of use above that consistent with good manufacturing practice.
(4) Where a flavouring preparation, other than a flavouring preparation that has been traditionally used in the variety, is added to a cheese as permitted in subsection (1), the words “with (naming the flavouring preparation)” shall be added to the common name on any label.
(5) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(6) Where a cheese is labelled as permitted in subsection (5), the word “smoked” shall be shown on the principal display panel.
TABLE
PART I
Item Column I Column II Column III Variety of Cheese Maximum percentage of moisture Minimum percentage of milk fat 1 Asiago 40.0 30.0 2 Baby Edam 47.0 21.0 3 Baby Gouda 45.0 26.0 4 Blue 47.0 27.0 5 Butter (Butterkäse) 46.0 27.0 6 Bra 36.0 26.0 7 Brick 42.0 29.0 8 Brie 54.0 23.0 9 Caciocavallo 45.0 24.0 10 Camembert (Carré de l’est) 56.0 22.0 11 Canadian Style Brick 42.0 29.0 12 Canadian Style Munster 46.0 27.0 13 Colby 42.0 29.0 14 Danbo 46.0 25.0 15 Edam 46.0 22.0 16 Elbo 46.0 25.0 17 Emmentaler (Emmental, Swiss) 40.0 27.0 18 Esrom 50.0 23.0 19 Farmer’s 44.0 27.0 20 Feta 55.0 22.0 21 Fontina 46.0 27.0 22 Fynbo 46.0 25.0 23 Gouda 43.0 28.0 24 Gournay 55.0 33.0 25 Gruyère 38.0 28.0 26 Havarti 50.0 23.0 27 Jack 50.0 25.0 28 Kasseri 44.0 25.0 29 Limburger 50.0 25.0 30 Maribo 43.0 26.0 31 Montasio 40.0 28.0 32 Monterey (Monterey Jack) 44.0 28.0 33 Mozzarella (Scamorza) 52.0 20.0 34 Muenster (Munster) 50.0 25.0 35 Neufchâtel 60.0 20.0 36 Parmesan 32.0 22.0 37 Part Skim Mozzarella (Part Skim Scamorza) 52.0 15.0 38 Part Skim Pizza 48.0 15.0 38.1 Part Skim Pizza Mozzarella 61.0 11.0 39 Pizza 48.0 20.0 39.1 Pizza Mozzarella 58.0 15.0 40 Provolone 45.0 24.0 41 Romano (Sardo) 34.0 25.0 42 St. Jorge 40.0 27.0 43 Saint-Paulin 50.0 25.0 44 Samsoë 44.0 26.0 45 Tilsiter (Tilsit) 45.0 25.0 46 Tybo 46.0 25.0 PART II
Item Column I Column II Column III Variety of Cheese Maximum percentage of moisture Maximum percentage of milk fat 1 Harzkase (Harzer Käse, Mainzer Käse) 55.0 3.0 2 Skim Milk 55.0 7.0
- SOR/79-752, s. 2
- SOR/80-632, s. 2
- SOR/82-383, ss. 2,3
- SOR/82-566, s. 1
- SOR/84-302, s. 1
- SOR/86-89, s. 2
- SOR/87-640, s. 3
- SOR/88-534, s. 2
- SOR/89-198, s. 1
- SOR/90-469, s. 1
- SOR/91-88, s. 1
- SOR/92-197, s. 1
- SOR/92-400, s. 4
- SOR/93-477, s. 1
- SOR/94-212, s. 1
- SOR/94-417, s. 1
- SOR/95-183, s. 1
- SOR/97-191, s. 1
- SOR/98-458, s. 2
- SOR/2000-336, s. 1
- SOR/2000-353, s. 4
- SOR/2000-417, s. 2
- SOR/2001-94, s. 1
- SOR/2005-98, s. 7
- SOR/2007-302, ss. 2, 4(F)
- SOR/2010-94, s. 8(E)
- SOR/2010-143, ss. 1, 39(E)
- SOR/2012-43, s. 2(F)
- SOR/2022-168, s. 52
B.08.034 (1) [S]. Cheddar Cheese
(a) shall
(i) be the product that is made by coagulating milk, milk products or a combination of those things with the aid of bacteria to form a curd and subjecting the curd to the cheddar process or any process other than the cheddar process that produces a cheese having the same physical, chemical and organoleptic properties as those of cheese produced by the cheddar process,
(i.1) have a casein content that is derived from milk or from ultrafiltered milk, partly skimmed milk, ultrafiltered partly skimmed milk, skim milk, ultrafiltered skim milk or cream, rather than from other milk products, that is at least 83 per cent of the total protein content of the cheese,
(i.2) have a whey protein to casein ratio that does not exceed the whey protein to casein ratio of milk, and
(ii) contain
(A) not more than 39 per cent moisture, and
(B) not less than 31 per cent milk fat;
(b) may contain
(i) salt,
(ii) flavouring preparations other than cheese flavouring,
(iii) bacterial cultures to aid further ripening,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8’-carotenal, ethyl beta-apo-8’-carotenoate,
(v) calcium chloride as a firming agent in an amount not exceeding 0.02 per cent of the milk and milk products used,
(vi) wood smoke as a preservative in an amount consistent with good manufacturing practice,
(vii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or a combination thereof in an amount not exceeding 2,000 parts per million calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate or a combination thereof in an amount not exceeding 3,000 parts per million calculated as sorbic acid,
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million calculated as propionic acid and sorbic acid respectively, or
(D) natamycin applied to the surface of the cheese in an amount that does not exceed 20 parts per million or, if the cheese is grated or shredded, 10 parts per million, and
(viii) in the case of grated or shredded cheddar cheese, calcium silicate, microcrystalline cellulose or cellulose, or a combination of them, as an anticaking agent, the total amount not to exceed 2.0 per cent; and
(c) shall not be labelled or advertised as cheddar cheese that has been aged unless
(i) it is made solely with milk, ultrafiltered milk, partly skimmed milk, ultrafiltered partly skimmed milk, skim milk, ultrafiltered skim milk or cream or a combination of those things, and
(ii) it has been aged for at least nine months and the period for which it has been aged is specified on the principal display panel of that label or in that advertising.
(1.1) Cheddar cheese may contain more than the maximum percentage of moisture set out in clause (1)(a)(ii)(A) and less than the minimum percentage of milk fat set out in clause (1)(a)(ii)(B) if
(a) a statement or claim set out in column 4 of any of items 12 to 14, 16, 20, 21 and 45 of the Table of Permitted Nutrient Content Statements and Claims is shown on the label of the product as part of the common name; and
(b) the cheese has the characteristic flavour and texture of cheddar cheese.
(1.2) The reference to “83 per cent” in subparagraph (1)(a)(i.1) shall be read as “78 per cent” if
(a) a statement or claim set out in column 4 of any of items 12 to 14, 16, 20, 21 and 45 of the Table of Permitted Nutrient Content Statements and Claims is shown on the label of the product as part of the common name; and
(b) the cheese has the characteristic flavour and texture of cheddar cheese.
(2) No person shall, in the manufacture of the cheddar cheese, use any enzyme other than
(a) aminopeptidase derived from Lactococcus lactis, chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS105), lipase derived from Animal pancreatic tissue; Aspergillus niger var.; Aspergillus oryzae var.; Edible forestomach tissue of calves, kids or lambs; Rhizopus oryzae var. or from Aspergillus oryzae (MLT-2) (pRML 787) (p3SR2); Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)); Rhizopus niveus, milk coagulating enzyme derived from Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)), from Mucor pusillus Lindt by pure culture fermentation process or from Aspergillus oryzae RET-1 (pBoel777), pepsin derived from glandular layer of porcine stomach, phospholipase derived from Aspergillus oryzae (pPFJo142) or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs;
(b) protease derived from Aspergillus oryzae; and
(c) lysozyme derived from egg-white.
(2.1) No person shall use an enzyme referred to in subsection (2) at a level of use above that consistent with good manufacturing practice.
(3) Where a flavouring preparation is added to a cheese as permitted in subsection (1), the words “with (naming the flavouring preparation)” shall be added to the common name in any label.
(4) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(5) Where a cheese is labelled as permitted in subsection (4), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/82-383, s. 4
- SOR/83-617, s. 1
- SOR/84-302, s. 2
- SOR/84-762, s. 7
- SOR/88-534, s. 3
- SOR/89-244, s. 1
- SOR/90-469, s. 2
- SOR/91-88, s. 2
- SOR/92-197, s. 2
- SOR/93-477, s. 2
- SOR/94-212, s. 2
- SOR/95-183, s. 2
- SOR/97-191, s. 2
- SOR/98-458, s. 3
- SOR/2000-336, s. 2
- SOR/2000-417, s. 3
- SOR/2005-98, s. 7
- SOR/2007-302, ss. 3, 4(F)
- SOR/2010-143, ss. 2, 39(E)
- SOR/2012-43, s. 3(F)
- SOR/2022-168, s. 52
B.08.035 (1) [S]. Cream Cheese
(a) shall
(i) be the product made by coagulating cream with the aid of bacteria to form a curd and forming the curd into a homogeneous mass after draining the whey, and
(ii) contain
(A) not more than 55 per cent moisture, and
(B) not less than 30 per cent milk fat; and
(b) may contain
(i) cream added to adjust the milk fat content,
(ii) salt,
(iii) nitrogen to improve spreadability in an amount consistent with good manufacturing practice,
(iv) the following emulsifying, gelling, stabilizing and thickening agents:
ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or any combination thereof in an amount not exceeding 0.5 per cent, and
(v) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) No person shall use any enzyme
(a) other than chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS 105), pepsin derived from glandular layer of porcine stomach or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in the manufacture of cream cheese; and
(b) at a level of use above that consistent with good manufacturing practice.
- SOR/79-752, s. 2
- SOR/92-197, s. 3
- SOR/94-212, s. 3
- SOR/95-183, s. 3
- SOR/2010-143, s. 3
B.08.036 (1) [S]. Whey Cheese or (naming the variety) Whey Cheese
(a) shall be the product made by coagulating whey or concentrated whey with the aid of heat to form a curd and shaping the curd; and
(b) may contain
(i) micro-organisms to aid further ripening,
(ii) added milk and milk products, and
(iii) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate, sodium hydroxide and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice.
(2) No person shall use, to aid coagulation of whey in the manufacture of whey cheese, a substance other than vinegar or sour whey.
- SOR/79-752, s. 2
- SOR/2007-302, s. 4(F)
- SOR/2010-142, s. 5
B.08.037 (1) [S]. Cream Cheese with (naming the added ingredients)
(a) shall
(i) be the product made by coagulating cream with the aid of bacteria to form a curd and forming the curd into a homogeneous mass after draining the whey, and
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from cream cheese but not in amounts so large as to change the basic nature of the product:
(A) cheese other than cream cheese,
(B) chocolate, condiments, flavouring preparations, seasonings or spices,
(C) fruits, nuts, pickles, relishes or vegetables,
(D) prepared or preserved meat, or
(E) prepared or preserved fish, and
(iii) contain
(A) not more than 60 per cent moisture, and
(B) not less than 26 per cent milk fat; and
(b) may contain
(i) cream added to adjust the milk fat content,
(ii) salt,
(iii) nitrogen to improve spreadability in an amount consistent with good manufacturing practice,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8’-carotenal, ethyl beta-apo-8’-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or any combination thereof in an amount not exceeding 0.5 per cent, and
(vi) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) No person shall use any enzyme
(a) other than chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS 105), pepsin derived from glandular layer of porcine stomach or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in the manufacture of a product to which subsection (1) applies; and
(b) at a level of use above that consistent with good manufacturing practice.
- SOR/79-752, s. 2
- SOR/92-197, s. 4
- SOR/94-212, s. 4
- SOR/95-183, s. 4
- SOR/2010-143, s. 4
- SOR/2011-278, s. 2
B.08.038 (1) [S]. Cream Cheese Spread
(a) shall
(i) be the product made by coagulating cream with the aid of bacteria to form a curd and forming the curd into a homogeneous mass after draining the whey, and
(ii) contain
(A) added milk and milk products,
(B) not less than 51 per cent cream cheese,
(C) not more than 60 per cent moisture, and
(D) not less than 24 per cent milk fat; and
(b) may contain
(i) cream added to adjust the milk fat content,
(ii) salt, vinegar and sweetening agents,
(iii) nitrogen to improve spreadability in an amount consistent with good manufacturing practice,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent, and
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice, and
(vii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) No person shall use any enzyme
(a) other than chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS 105), pepsin derived from glandular layer of porcine stomach or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in the manufacture of a product to which subsection (1) applies; and
(b) at a level of use above that consistent with good manufacturing practice.
- SOR/79-752, s. 2
- SOR/92-197, s. 5
- SOR/94-212, s. 5
- SOR/95-183, s. 5
- SOR/2007-302, s. 4(F)
- SOR/2010-143, s. 5
B.08.039 (1) [S]. Cream Cheese Spread with (naming the added ingredients)
(a) shall
(i) be the product made by coagulating cream with the aid of bacteria to form a curd and forming the curd into a homogeneous mass after draining the whey,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from cream cheese spread but not in amounts so large as to change the basic nature of the product:
(A) cheese other than cream cheese,
(B) chocolate, condiments, flavouring preparations, seasonings or spices,
(C) fruits, nuts, pickles, relishes or vegetables,
(D) prepared or preserved meat, or
(E) prepared or preserved fish, and
(iii) contain
(A) added milk and milk products,
(B) not more than 60 per cent moisture, and
(C) not less than 24 per cent milk fat; and
(b) may contain
(i) cream added to adjust the milk fat content,
(ii) salt, vinegar and sweetening agents,
(iii) nitrogen to improve spreadability in an amount consistent with good manufacturing practice,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric,
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate, and
(C) in an amount not exceeding 1.5%, caramel,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent, and
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice, and
(vii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) No person shall use any enzyme
(a) other than chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS 105), pepsin derived from glandular layer of porcine stomach or rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in the manufacture of a product to which subsection (1) applies; and
(b) at a level of use above that consistent with good manufacturing practice.
- SOR/79-752, s. 2
- SOR/92-197, s. 6
- SOR/94-212, s. 6
- SOR/95-183, s. 6
- SOR/2007-302, s. 4(F)
- SOR/2010-143, s. 6
- SOR/2011-278, s. 3
- SOR/2011-281, s. 1
B.08.040 (1) [S]. Processed (naming the variety) Cheese
(a) shall
(i) subject to subparagraph (ii), be the product made by comminuting and mixing the named variety or varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat,
(ii) in the case of processed cheddar cheese, be the product made by comminuting and mixing one or more of the following:
(A) cheddar cheese,
(B) stirred curd cheese,
(C) granular curd cheese, or
(D) washed curd cheese
into a homogeneous mass with the aid of heat,
(iii) have, where it is made from
(A) one variety of cheese, in which the maximum amount of moisture permitted is less than 40 per cent, or
(B) two or more varieties of cheese, in which the average maximum amount of moisture permitted is less than 40 per cent,
a moisture content that does not exceed, by more than five per cent, the amount referred to in clause (A) or (B), as the case may be, and a milk fat content that is not less, by more than three per cent, than the minimum milk fat content or average minimum milk fat content permitted for that variety or those varieties, as the case may be,
(iv) subject to subparagraph (v), have, where it is made from
(A) one variety of cheese, in which the maximum amount of moisture permitted is 40 per cent or more, or
(B) more than one variety of cheese, in which the average maximum amount of moisture permitted is 40 per cent or more,
a moisture content that does not exceed, by more than three per cent, the amount referred to in clause (A) or (B), as the case may be, and a milk fat content that is not less, by more than two per cent, than the minimum milk fat content or average minimum milk fat content permitted for that variety or those varieties, as the case may be, and
(v) in the case of processed skim milk cheese, contain not more than
(A) 55 per cent moisture, and
(B) seven per cent milk fat; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) in the case of processed skim milk cheese, added skim milk powder, buttermilk powder and whey powder,
(iv) salt, vinegar and sweetening agents,
(v) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(vi) the following emulsifying, gelling, stabilizing and thickening agents:
(A) sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate) in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(vi.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vii) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(viii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(ix) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/91-409, s. 1
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041 (1) [S]. Processed (naming the variety) Cheese with (naming the added ingredients)
(a) shall
(i) be the product made by comminuting and mixing the named variety or varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from processed (naming the variety) cheese but not in amounts so large as to change the basic nature of the product:
(A) flavouring preparations other than such preparations that resemble the flavour of the varieties of cheese used in the product,
(B) chocolate, condiments, seasonings or spices,
(C) fruits, nuts, pickles, relishes or vegetables,
(D) prepared or preserved meat, or
(E) prepared or preserved fish,
(iii) have, where it is made from
(A) one variety of cheese, in which the maximum amount of moisture permitted is less than 40 per cent, or
(B) more than one variety of cheese, in which the average maximum amount of moisture permitted is less than 40 per cent,
a moisture content that does not exceed by more than five per cent, the amount referred to in clause (A) or (B), as the case may be, and a milk fat content that is not less, by more than three per cent, than the minimum milk fat content or average minimum milk fat content permitted for that variety or those varieties, as the case may be, and
(iv) have, where it is made from
(A) one variety of cheese, in which the maximum amount of moisture permitted is 40 per cent or more, or
(B) more than one variety of cheese, in which the average maximum amount of moisture permitted is 40 per cent or more,
a moisture content that does not exceed, by more than three per cent, the amount referred to in clause (A) or (B), as the case may be, and a milk fat content that is not less, by more than two per cent, than the minimum milk fat content or average minimum milk fat content permitted for that variety or those varieties, as the case may be; and
(b) may contain
(i) water added to adjust moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate) in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(v.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/91-409, s. 2
- SOR/92-400, s. 5
- SOR/2010-94, s. 4(E)
- SOR/2011-278, s. 4
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.1 (1) [S]. Processed Cheese Food
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat, and
(ii) contain
(A) added milk or milk products,
(B) not less than 51 per cent cheese,
(C) not more than 46 per cent moisture, and
(D) not less than 23 per cent milk fat; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate) in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(v.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/91-409, s. 3
- SOR/92-400, s. 6
- SOR/2007-302, s. 4(F)
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.2 (1) [S]. Processed Cheese Food with (naming the added ingredients)
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from processed cheese food but not in amounts so large as to change the basic nature of the product:
(A) chocolate, condiments, flavouring preparations, seasonings or spices,
(B) fruits, nuts, pickles, relishes or vegetables,
(C) prepared or preserved meat, or
(D) prepared or preserved fish, and
(iii) contain
(A) added milk or milk products,
(B) not more than 46 per cent moisture, and
(C) not less than 22 per cent milk fat; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8’-carotenal, ethyl beta-apo-8’-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate) in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(v.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/91-409, s. 4
- SOR/92-400, s. 7
- SOR/2007-302, s. 4(F)
- SOR/2011-278, s. 5
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.3 (1) [S]. Processed Cheese Spread
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat, and
(ii) contain
(A) added milk or milk products,
(B) not less than 51 per cent cheese,
(C) not more than 60 per cent moisture, and
(D) not less than 20 per cent milk fat;
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8’-carotenal, ethyl beta-apo-8’-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(v.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/82-1071, s. 4
- SOR/91-409, s. 5
- SOR/2007-302, s. 4(F)
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.4 (1) [S]. Processed Cheese Spread with (naming the added ingredients)
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass with the aid of heat,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from processed cheese spread but not in amounts so large as to change the basic nature of the product:
(A) chocolate, condiments, flavouring preparations, seasonings or spices,
(B) fruits, nuts, pickles, relishes or vegetables,
(C) prepared or preserved meat, or
(D) prepared or preserved fish, and
(iii) contain
(A) added milk or milk products,
(B) not more than 60 per cent moisture, and
(C) not less than 20 per cent milk fat; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
(A) ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent,
(B) calcium phosphate dibasic, potassium phosphate dibasic, sodium acid pyrophosphate, sodium aluminum phosphate, sodium hexametaphosphate, sodium phosphate dibasic, sodium phosphate monobasic, sodium phosphate tribasic, sodium pyrophosphate tetrabasic, calcium citrate, potassium citrate, sodium citrate, sodium potassium tartrate, sodium tartrate, sodium gluconate or any combination thereof in an amount, when calculated as anhydrous salts, not exceeding 3.5 per cent in the case of phosphate salts and 4.0 per cent in total,
(C) lecithin in an amount not exceeding 0.2 per cent, and
(D) monoglycerides, mono- and diglycerides or any combination thereof in an amount not exceeding 0.5 per cent,
(v.1) calcium phosphate tribasic as an agent to improve colour, texture, consistency and spreadability, in an amount not exceeding 1 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/82-1071, s. 5
- SOR/91-409, s. 6
- SOR/2007-302, s. 4(F)
- SOR/2011-278, s. 6
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.5 (1) [S]. Cold-Pack (naming the variety) Cheese
(a) shall
(i) subject to subparagraph (ii), be the product made by comminuting and mixing the named variety or varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass without the aid of heat,
(ii) in the case of cold-pack cheddar cheese, be the product made by comminuting and mixing one or more of the following:
(A) cheddar cheese,
(B) stirred curd cheese,
(C) granular curd cheese, or
(D) washed curd cheese
into a homogeneous mass without the aid of heat,
(iii) contain, where it is made from
(A) one variety of cheese, not more moisture and not less milk fat than the maximum moisture content and minimum fat content permitted for that variety, or
(B) more than one variety of cheese, not more moisture and not less milk fat than the average maximum moisture content and the average minimum fat content permitted for those varieties; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vi) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(vii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/92-400, s. 8
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.6 (1) [S]. Cold-Pack (naming the variety) Cheese with (naming the added ingredients)
(a) shall
(i) be the product made by comminuting and mixing the named variety or varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass without the aid of heat,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from cold-pack (naming the variety) cheese but not in amounts so large as to change the basic nature of the product:
(A) flavouring preparations other than such preparations that resemble the flavour of the varieties of cheese used in the product,
(B) chocolate, condiments, seasonings or spices,
(C) fruits, nuts, pickles, relishes or vegetables,
(D) prepared or preserved meat, or
(E) prepared or preserved fish, and
(iii) contain, where it is made from
(A) one variety of cheese, not more moisture and not less milk fat than the maximum moisture content and one per cent less than the minimum milk fat content permitted for that variety, or
(B) more than one variety of cheese, not more moisture and not less milk fat than the average maximum moisture content and one per cent less than the average minimum milk fat content permitted for those varieties; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/92-400, s. 9
- SOR/2010-94, s. 5(E)
- SOR/2011-278, s. 7
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.7 (1) [S]. Cold-Pack Cheese Food
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass without the aid of heat,
(ii) contain
(A) added milk or milk products,
(B) not less than 51 per cent cheese,
(C) not more than 46 per cent moisture, and
(D) not less than 23 per cent milk fat; and
(b) may contain
(i) water added to adjust the moisture content,
(ii) added milk fat,
(iii) salt, vinegar and sweetening agents,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/2007-302, s. 4(F)
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.041.8 (1) [S]. Cold-Pack Cheese Food with (naming the added ingredients)
(a) shall
(i) be the product made by comminuting and mixing one or more varieties of cheese, other than cream cheese, cottage cheese or whey cheese, into a homogeneous mass without the aid of heat,
(ii) contain the named added ingredients which shall be one or more of the following ingredients in amounts sufficient to differentiate the product from cold-pack cheese food but not in amounts so large as to change the basic nature of the product:
(A) chocolate, condiments, flavouring preparations, seasonings or spices,
(B) fruits, nuts, pickles, relishes or vegetables,
(C) prepared or preserved meat, or
(D) prepared or preserved fish, and
(iii) contain
(A) added milk or milk products,
(B) not more than 46 per cent moisture, and
(C) not less than 22 per cent milk fat; and
(b) may contain
(i) water added to adjust moisture content,
(ii) added milk fat,
(iii) sweetening agents, salt and vinegar,
(iv) one or more of the following colouring agents:
(A) in an amount consistent with good manufacturing practice, annatto, beta-carotene, chlorophyll, paprika, riboflavin, turmeric, and
(B) in an amount not exceeding 35 parts per million, either singly or in combination thereof, beta-apo-8′-carotenal, ethyl beta-apo-8′-carotenoate,
(v) the following emulsifying, gelling, stabilizing and thickening agents:
ammonium carrageenan, calcium carrageenan, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, Irish Moss Gelose, potassium carrageenan, propylene glycol alginate, sodium carboxymethyl cellulose (carboxymethyl cellulose, cellulose gum, sodium cellulose glycolate), sodium carrageenan, tragacanth gum, xanthan gum or a combination thereof in an amount not exceeding 0.5 per cent,
(vi) acetic acid, calcium carbonate, citric acid, lactic acid, malic acid, phosphoric acid, potassium bicarbonate, potassium carbonate, sodium bicarbonate, sodium carbonate and tartaric acid as pH adjusting agents in an amount consistent with good manufacturing practice,
(vii) wood smoke as a preservative in an amount consistent with good manufacturing practice, and
(viii) the following preservatives:
(A) propionic acid, calcium propionate, sodium propionate or any combination thereof in an amount not exceeding 2,000 parts per million, calculated as propionic acid,
(B) sorbic acid, calcium sorbate, potassium sorbate, sodium sorbate, or any combination thereof in an amount not exceeding 3,000 parts per million, calculated as sorbic acid, or
(C) any combination of the preservatives named in clauses (A) and (B) in an amount not exceeding 3,000 parts per million, calculated as propionic acid and sorbic acid respectively.
(2) Only a cheese to which wood smoke has been added as permitted in subsection (1) may be described by the term “smoked” on a label.
(3) Where a cheese is labelled as permitted in subsection (2), the word “smoked” shall be shown on the principal display panel.
- SOR/79-752, s. 2
- SOR/2007-302, s. 4(F)
- SOR/2011-278, s. 8
- SOR/2017-18, s. 18(F)
- SOR/2018-69, s. 28(F)
B.08.042 No manufacturer shall sell whole cheese that is not made from a pasteurized source unless the date of the beginning of the manufacturing process is
(a) marked or branded thereon within three days thereof; or
(b) marked on the label at the time of packaging, if the cheese is such that, because of its texture, consistency, or physical structure, such date cannot be effectively branded or marked on the cheese.
B.08.043 No manufacturer shall sell any cheese that is not made from a pasteurized source if it has been cut into smaller portions, unless
(a) it has been duly stored; or
(b) each portion of cut cheese is marked, branded or labelled with the date of the beginning of the manufacturing process.
B.08.044 (1) Subject to subsection (2), no person shall sell cheese, including cheese curd, that is not made from a pasteurized source unless it has been stored.
(2) Cheese, including cheese curd, that is not made from a pasteurized source may be used as an ingredient in any food providing such food is manufactured or processed so as to pasteurize the cheese in the manner described in the definition pasteurized source in section B.08.030(1).
- SOR/78-405, s. 1
- SOR/79-752, s. 3
B.08.045 Notwithstanding B.08.044, cheese that has not been manufactured from a pasteurized source and has not been stored but is marked or branded with the date of the beginning of the manufacturing process, may be sold to
(a) a wholesaler;
(b) a jobber; or
(c) in quantities of not less than 900 pounds, to a retailer.
B.08.046 No person shall sell any whole cheese that has not been made from a pasteurized source unless there is stamped thereon the date of the beginning of the manufacturing process.
B.08.047 Every manufacturer, wholesaler, or jobber who sells cheese not made from a pasteurized source and which has not been stored shall keep a record of
(a) the registered number of the cheese factory,
(b) the date of manufacture of the cheese,
(c) the vat number or vat numbers,
(d) the name and address of the person to whom the cheese is sold, and
(e) the weight sold from each vat,
for each lot of cheese sold.
B.08.048 (1) Subject to section B.08.054, no person shall sell cheese, including cheese curd, made from a pasteurized source if the cheese contains more than
(a) 100 Escherichia coli, or
(b) 100 Staphylococcus aureus
per gram, as determined by official method MFO-14, Microbiological Examination of Cheese, November 30, 1983.
(2) No person shall sell cheese, made from an unpasteurized source if the cheese contains more than
(a) 500 Escherichia coli, or
(b) 1,000 Staphylococcus aureus
per gram, as determined by official method MFO-14, Microbiological Examination of Cheese, November 30, 1983.
- SOR/78-405, s. 2
- SOR/82-768, s. 21
- SOR/84-17, s. 4
B.08.049 [S]. Whey
(a) shall be the product remaining after the curd has been removed from milk in the process of making cheese; and
(b) may contain
(i) catalase, in the case of liquid whey that has been treated with hydrogen peroxide,
(ii) lactase,
(iii) hydrogen peroxide, in the case of liquid whey destined for the manufacture of dried whey products,
(iv) benzoyl peroxide, and calcium phosphate tribasic and calcium sulphate as carriers of the benzoyl peroxide, in the case of liquid whey destined for the manufacture of dried whey products other than those for use in infant formula, and
(v) sodium hexametaphosphate, in the case of liquid whey destined for the manufacture of concentrated or dried whey products.
- SOR/79-752, s. 4
- SOR/89-555, s. 1
- SOR/2010-40, s. 1
- SOR/2011-282, s. 1
B.08.050 [Repealed, SOR/95-281, s. 1]
B.08.051 [S]. Cottage Cheese
(a) shall be the product, in the form of discrete curd particles, prepared from skim milk, evaporated skim milk or skim milk powder and harmless acid-producing bacterial cultures;
(b) shall contain not more than 80 per cent moisture;
(c) may contain not more than 0.5 per cent stabilizing agent; and
(d) may contain
(i) milk,
(ii) cream,
(iii) milk powder,
(iv) rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs,
(v) a milk coagulating enzyme derived from Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)), from Mucor pusillus Lindt by pure culture fermentation process or from Aspergillus oryzae RET-1 (pBoel777), in an amount consistent with good manufacturing practice,
(vi) chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), in an amount consistent with good manufacturing practice,
(vi.1) chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS105), in an amount consistent with good manufacturing practice,
(vi.2) pepsin derived from glandular layer of porcine stomach,
(vii) salt,
(viii) calcium chloride,
(ix) added lactose,
(x) pH adjusting agents,
(xi) relishes,
(xii) fruits,
(xiii) vegetables, and
(xiv) carbon dioxide.
- SOR/81-60, s. 4
- SOR/92-197, s. 7
- SOR/94-212, s. 7
- SOR/95-183, s. 7
- SOR/98-458, s. 4
- SOR/2001-94, s. 2
- SOR/2005-98, s. 7
- SOR/2010-143, ss. 7, 39(E)
B.08.052 [S]. Creamed Cottage Cheese shall be cottage cheese containing cream or a mixture of cream with milk or skim milk, or both, in such quantity that the final product shall contain
(a) not less than four per cent milk fat; and
(b) not more than 80 per cent moisture and may contain emulsifying, gelling, stabilizing and thickening agents.
B.08.053 All dairy products used in the preparation of cottage cheese shall be from a pasteurized source.
B.08.054 No person shall sell cottage cheese or creamed cottage cheese that contains more than 10 coliform bacteria per gram, as determined by official method MFO-4, Microbiological Examination of Cottage Cheese, November 30, 1981.
- SOR/82-768, s. 22
Butter
B.08.056 [S]. Butter
(a) shall
(i) be the food prepared in accordance with good manufacturing practices from milk or milk products, and
(ii) contain not less than 80 per cent milk fat; and
(b) may contain
(i) milk solids,
(ii) bacterial culture,
(iii) salt, and
(iv) food colour.
- SOR/92-400, s. 10
B.08.057 [S]. Whey Butter shall be butter made from whey cream.
- SOR/92-400, s. 10
Ice Cream
B.08.061 [S]. Ice Cream Mix
(a) shall be the unfrozen, pasteurized combination of cream, milk or other milk products, sweetened with sugar, liquid sugar, invert sugar, honey, dextrose, glucose, corn syrup, corn syrup solids or any combination of such sweeteners;
(b) may contain
(i) egg,
(ii) a flavouring preparation,
(iii) cocoa or chocolate syrup,
(iv) a food colour,
(v) pH adjusting agents,
(vi) microcrystalline cellulose or a stabilizing agent or both in an amount that will not exceed 0.5 per cent of the ice cream made from the mix,
(vii) a sequestering agent,
(viii) salt,
(ix) not more than one per cent added edible casein or edible caseinates; and
(x) propylene glycol mono fatty acid esters in an amount that will not exceed 0.35 per cent of the ice cream made from the mix and sorbitan tristearate in an amount that will not exceed 0.035 per cent of the ice cream made from the mix; and
(c) shall contain not less than
(i) 36 per cent solids, and
(ii) 10 per cent milk fat or, where cocoa or chocolate syrup has been added, eight per cent milk fat.
- SOR/92-400, s. 11
- SOR/97-543, s. 2(F)
- SOR/2007-75, s. 2
- SOR/2007-302, s. 4(F)
- SOR/2010-142, s. 6(F)
B.08.062 [S]. Ice Cream
(a) shall be the frozen food obtained by freezing an ice cream mix, with or without the incorporation of air;
(b) may contain cocoa or chocolate syrup, fruit, nuts or confections;
(c) shall contain not less than
(i) 36 per cent solids,
(ii) 10 per cent milk fat, or, where cocoa or chocolate syrup, fruit, nuts, or confections have been added, eight per cent milk fat, and
(iii) 180 grams of solids per litre of which amount not less than 50 grams shall be milk fat, or, where cocoa or chocolate syrup, fruit, nuts or confections have been added, 180 grams of solids per litre of which amount not less than 40 grams shall be milk fat; and
(d) shall contain not more than
(i) 100,000 bacteria per gram, and
(ii) 10 coliform organisms per gram,
as determined by official method MFO-2, Microbiological Examination of Ice Cream or Ice Milk, November 30, 1981.
- SOR/82-768, s. 23
- SOR/92-400, s. 12
Sherbet
- SOR/98-580, s. 1(F)
B.08.063 [S]. Sherbet
(a) shall be the frozen food, other than ice cream or ice milk, made from a milk product;
(b) may contain
(i) water,
(ii) a sweetening agent,
(iii) fruit or fruit juice,
(iv) citric or tartaric acid,
(v) a flavouring preparation,
(vi) a food colour,
(vii) not more than 0.75 per cent stabilizing agent,
(viii) a sequestering agent,
(ix) lactose,
(x) not more than 0.5 per cent microcrystalline cellulose, and
(xi) not more than one per cent added edible casein or edible caseinates; and
(c) shall contain
(i) not more than five per cent milk solids, including milk fat, and
(ii) not less than 0.35 per cent acid determined by titration and expressed as lactic acid.
- SOR/92-400, s. 13
- SOR/97-543, s. 3(F)
- SOR/98-580, s. 1(F)
- SOR/2007-302, s. 4(F)
Ice Milk
B.08.071 [S]. Ice Milk Mix
(a) shall be the unfrozen, pasteurized combination of cream, milk or other milk products, sweetened with sugar, liquid sugar, invert sugar, honey, dextrose, glucose, corn syrup, corn syrup solids or any combination of such sweeteners;
(b) may contain
(i) egg,
(ii) a flavouring preparation,
(iii) cocoa or chocolate syrup,
(iv) a food colour,
(v) a pH adjusting agent,
(vi) a stabilizing agent, in an amount that will not result in more than 0.5 per cent stabilizing agent in the ice milk,
(vii) a sequestering agent,
(viii) added lactose, and
(ix) not more than 1.5 per cent microcrystalline cellulose,
(x) salt, and
(xi) not more than one per cent added edible casein or edible caseinates; and
(c) shall contain
(i) not less than 33 per cent solids, and
(ii) not less than three per cent and not more than five per cent milk fat.
- SOR/92-400, s. 14
- SOR/97-543, s. 4(F)
- SOR/2007-302, s. 4(F)
B.08.072 [S]. Ice Milk
(a) shall be the frozen food obtained by freezing an ice milk mix, with or without the incorporation of air;
(b) may contain cocoa or chocolate syrup, fruit, nuts or confections;
(c) shall contain
(i) not less than 33 per cent solids,
(ii) not less than three per cent and not more than five per cent milk fat, and
(iii) not less than 160 grams of solids per litre of which amount not less than 14 grams shall be milk fat; and
(d) shall contain not more than
(i) 100,000 bacteria per gram, and
(ii) 10 coliform organisms per gram,
as determined by official method MFO-2, Microbiological Examination of Ice Cream or Ice Milk, November 30, 1981.
- SOR/82-768, s. 24
- SOR/92-400, s. 15
B.08.073 [Repealed, SOR/92-626, s. 13]
B.08.074 (1) The percentage of milk fat contained in
(a) yogurt,
(b) cottage cheese, and
(c) creamed cottage cheese,
shall be shown on the principal display panel followed by the words “milk fat” or the abbreviation “B.F.” or “M.F.”.
(2) In addition to the statement referred to in subsection (1), a person may, on the label of a food referred to in subsection (1), make a declaration of the fat content of the food, expressed in grams per serving of stated size.
- SOR/88-559, s. 18
- SOR/2016-305, s. 75(F)
Cream
B.08.075 [S]. Cream
(a) shall be the fatty liquid prepared from milk by separating the milk constituents in such a manner as to increase the milk fat content; and
(b) may contain
(i) a pH adjusting agent,
(ii) a stabilizing agent,
(iii) in the case of cream for whipping that has been heat-treated above 100°C, the following ingredients and food additives:
(A) skim milk powder in an amount not exceeding 0.25 per cent,
(B) glucose solids in an amount not exceeding 0.1 per cent,
(C) calcium sulphate in an amount not exceeding 0.005 per cent, and
(D) xanthan gum in an amount not exceeding 0.02 per cent, and
(E) [Repealed, SOR/2010-142, s. 7]
(iv) in the case of cream for whipping, microcrystalline cellulose in an amount not exceeding 0.2 per cent.
- SOR/79-662, s. 2
- SOR/82-1071, s. 6
- SOR/88-419, s. 1
- SOR/2010-142, s. 7
B.08.076 (1) The percentage of milk fat contained in canned cream shall be shown on the principal display panel followed by the words “milk fat” or the abbreviation “B.F.” or “M.F.”.
(2) In addition to the statement referred to in subsection (1), a person may, on the label of canned cream, make a declaration of the fat content of the cream, expressed in grams per serving of stated size.
- SOR/88-559, s. 19
- SOR/2016-305, s. 75(F)
B.08.077 [S]. Sour Cream
(a) shall be the product prepared by the souring of pasteurized cream with acid-producing bacterial culture and shall contain not less than 14 per cent milk fat; and
(b) may contain
(i) milk solids,
(ii) whey solids,
(iii) buttermilk,
(iv) starch in an amount not exceeding one per cent,
(v) salt,
(vi) rennet derived from aqueous extracts from the fourth stomach of calves, kids or lambs, in an amount consistent with good manufacturing practice,
(vii) the following emulsifying, gelling, stabilizing and thickening agents:
(A) algin, carob bean gum (locust bean gum), carrageenan, gelatin, guar gum, pectin or propylene glycol alginate or any combination thereof in an amount not exceeding 0.5 per cent,
(B) monoglycerides, mono- and diglycerides, or any combination thereof, in an amount not exceeding 0.3 per cent, and
(C) sodium phosphate dibasic in an amount not exceeding 0.05 per cent,
(viii) sodium citrate as a flavour precursor in an amount not exceeding 0.1 per cent,
(ix) a milk coagulating enzyme derived from Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)), from Mucor pusillus Lindt by pure culture fermentation process or from Aspergillus oryzae RET-1 (pBoel777), in an amount consistent with good manufacturing practice,
(x) chymosin A derived from Escherichia coli K-12, GE81 (pPFZ87A), in an amount consistent with good manufacturing practice, and
(xi) chymosin B derived from Aspergillus niger var. awamori, GCC0349 (pGAMpR) or from Kluyveromyces marxianus var. lactis, DS1182 (pKS105), in an amount consistent with good manufacturing practice.
- SOR/78-876, s. 1
- SOR/80-500, s. 3
- SOR/81-60, s. 5
- SOR/92-197, s. 8
- SOR/94-212, s. 8
- SOR/95-183, s. 8
- SOR/98-458, s. 5
- SOR/2005-98, s. 7
- SOR/2010-143, ss. 8, 39(E)
DIVISION 9Fats and Oils
B.09.001 [S]. Vegetable fats and oils shall be fats and oils obtained entirely from the botanical source after which they are named, shall be dry and sweet in flavour and odour and, with the exception of olive oil, may contain emulsifying agents, Class IV preservatives, an antifoaming agent, and B-carotene in a quantity sufficient to replace that lost during processing, if such an addition is declared on the label.
- SOR/85-179, s. 1
B.09.002 [S]. Animal fats and oils shall be fats and oils obtained entirely from animals healthy at the time of slaughter, shall be dry and sweet in flavour and odour and may contain
(a) with the exception of milk fat and suet, Class IV preservatives; and
(b) with the exception of lard, milk fat and suet, an antifoaming agent.
B.09.003 [S]. Olive Oil or Sweet Oil
(a) shall be the oil obtained from the fruit of the olive tree (Olea europaea L);
(b) shall have a fatty acid content that is
(i) not less than 56.0 and not more than 83.0 per cent oleic acid,
(ii) not less than 7.5 and not more than 20.0 per cent palmitic acid,
(iii) not less than 3.5 and not more than 20.0 per cent linoleic acid,
(iv) not less than 0.5 and not more than 3.5 per cent stearic acid,
(v) not less than 0.3 and not more than 3.5 per cent palmitoleic acid,
(vi) not more than 1.5 per cent linolenic acid, and
(vii) not more than 0.05 per cent myristic acid, calculated as methyl esters;
(c) shall not contain more than minute amounts of arachidic acid, behenic acid, gadoleic acid or lignoceric acid;
(d) shall have
(i) a relative density of not less than 0.910 and not more than 0.916, calculated with the oil at 20°C and water at 20°C (20°C/water at 20°C),
(ii) a refractive index of not less than 1.4677 and not more than 1.4705, calculated using the sodium D-line as the light source and with the oil at 20°C (nD20°C),
(iii) an iodine value of not less than 75 and not more than 94, calculated using the Wijs test,
(iv) a saponification value of not less than 184 and not more than 196, expressed as milligrams of potassium hydroxide per gram of oil,
(v) an acid value of not more than 6.6 milligrams potassium hydroxide per gram of oil,
(vi) a free acidity of not more than 3.3 per cent expressed as oleic acid,
(vii) a peroxide value of not more than 20 milliequivalents peroxide oxygen per kilogram of oil,
(viii) an unsaponifiable matter content of not more than 15 grams per kilogram, and
(ix) a Bellier index of not more than 17;
(e) shall show negative results when tested for semi-siccative oil, olive-residue oil, cotton-seed oil, tea-seed oil or sesame seed oil; and
(f) notwithstanding section B.09.001, may contain alpha-tocopherol in a quantity sufficient to replace that lost during refining, if such an addition is declared on the label.
- SOR/78-655, s. 1
B.09.004 [S]. Cotton Seed Oil
(a) shall be the oil of seeds of cultivated Gossypium spp.;
(b) shall have
(i) a relative density (20°C/water at 20°C) of not less than 0.918 and not more than 0.926,
(ii) a refractive index (nD40°C) of not less than 1.458 and not more than 1.466,
(iii) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 189 and not more than 198,
(iv) an iodine value (Wijs) of not less than 99 and not more than 119,
(v) an unsaponifiable matter content of not more than 15 grams per kilogram,
(vi) a positive Halphen test,
(vii) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(viii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil; and
(c) may contain oxystearin.
B.09.005 [S]. Cocoa Butter shall be the fat from cocoa nibs, obtained either before or after roasting, or cocoa liquor, and shall have
(a) a refractive index (40°C) of not less than 1.453 and not more than 1.458;
(b) a saponification value of not less than 188 and not more than 202;
(c) an iodine value (Hanus) of not less than 32 and not more than 41; and
(d) an acid value of not more than five.
- SOR/97-263, s. 3
B.09.006 [S]. Corn Oil or Maize Oil
(a) shall be the oil of the germ or embryo of Zea mays L.; and
(b) shall have
(i) a relative density of (20°C/water at 20°C) of not less than 0.917 and not more than 0.925,
(ii) a refractive index (nD40°C) of not less than 1.465 and not more than 1.468,
(iii) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 187 and not more than 195,
(iv) an iodine value (Wijs) of not less than 103 and not more than 128,
(v) an unsaponifiable matter content of not more than 28 grams per kilogram,
(vi) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(vii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil.
B.09.007 [S]. Peanut Oil or Arachis Oil
(a) shall be the oil of the seeds of Arachis hypogaea L.;
(b) shall have
(i) a relative density of (20°C/water at 20°C) of not less than 0.914 and not more than 0.917,
(ii) a refractive index (nD40°C) of not less than 1.460 and not more than 1.465,
(iii) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 187 and not more than 196,
(iv) an iodine value (Wijs) of not less than 80 and not more than 106,
(v) an unsaponifiable matter content of not more than 10 grams per kilogram,
(vi) an arachidic and higher fatty acids content of not less than 48 grams per kilogram,
(vii) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(viii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil; and
(c) not contain oxystearin.
- SOR/84-300, s. 22(E)
B.09.008 [S]. Soybean Oil, Soya Bean Oil, Soja Oil or Soya Oil
(a) shall be the oil of the seeds of Glycine max (L.) Merr.;
(b) shall have
(i) a relative density of (20°C/water at 20°C) of not less than 0.919 and not more than 0.925,
(ii) a refractive index (nD40°C) of not less than 1.466 and not more than 1.470,
(iii) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 189 and not more than 195,
(iv) an iodine value (Wijs) of not less than 120 and not more than 143,
(v) an unsaponifiable matter content of not more than 15 grams per kilogram,
(vi) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(vii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil; and
(c) may contain oxystearin.
- SOR/84-300, s. 23(E)
B.09.009 [S]. Sunflowerseed Oil or Sunflower Oil
(a) shall be the oil of the seeds of Helianthus annuus L.; and
(b) shall have
(i) a relative density of (20°C/water at 20°C) of not less than 0.918 and not more than 0.923,
(ii) a refractive index (nD40°C) of not less than 1.467 and not more than 1.469,
(iii) an iodine value (Wijs) of not less than 110 and not more than 143,
(iv) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 188 and not more than 194,
(v) an unsaponifiable matter content of not more than 15 grams per kilogram,
(vi) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(vii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil.
B.09.009A [S]. Safflowerseed Oil or Safflower Oil
(a) shall be the oil of the seeds of Carthamus tinctorius L.;
(b) shall have
(i) a relative density of (20°C/water at 20°C) of not less than 0.922 and not more than 0.927,
(ii) a refractive index (nD40°C) of not less than 1.467 and not more than 1.470,
(iii) a saponification value (milligrams potassium hydroxide per gram of oil) of not less than 186 and not more than 198,
(iv) an iodine value (Wijs) of not less than 135 and not more than 150,
(v) an unsaponifiable matter content of not more than 15 grams per kilogram,
(vi) an acid value of not more than 0.6 milligram potassium hydroxide per gram of oil, and
(vii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of oil.
B.09.010 Despite the common name set out in the Common Names for Ingredients and Components Document, if a vegetable fat or oil is an ingredient of any cooking oil, salad oil or table oil, the fat or oil shall be shown in the list of ingredients by its common name.
- SOR/98-458, s. 7(F)
- SOR/2022-143, s. 20
B.09.011 [S]. Shortening, other than butter or lard, shall be the semi-solid food prepared from fats, oils or a combination of fats and oils, may be processed by full hydrogenation and may contain
(a) Class IV preservatives,
(b) an antifoaming agent,
(c) stearyl monoglyceridyl citrate,
(d) monoglycerides or a combination of monoglycerides and diglycerides of fat forming fatty acids, the weight of the monoglycerides being not more than 10 per cent and the total weight of monoglycerides and diglycerides being not more than 20 per cent of the weight of the shortening,
(e) lactylated monoglycerides, or a combination of lactylated monoglycerides and diglycerides of fat forming fatty acids, the total weight being not more than eight per cent of the weight of the shortening, and
(f) sorbitan tristearate,
except that the total weight of the ingredients permitted under paragraphs (d) and (e) shall not be greater than 20 per cent of the weight of the shortening.
B.09.012 [Repealed, SOR/97-148, s. 2]
B.09.013 [S]. Lard
(a) shall be the rendered fat from hogs;
(b) shall have
(i) a relative density of not less than 0.894 and not more than 0.906, calculated with the lard at 40°C and water at 20°C (40°C/water at 20°C),
(ii) a refractive index of not less than 1.448 and not more than 1.461, calculated using the sodium D-line as the light source and with the lard at 40°C (nD40°C),
(iii) a titre of not less than 32°C and not more than 45°C,
(iv) a saponification value of not less than 192 and not more than 203, expressed as milligrams potassium hydroxide per gram of fat,
(v) an iodine value of not less than 45 and not more than 70, calculated using the Wijs test,
(vi) an unsaponifiable matter content of not more than 12 grams per kilogram,
(vii) an acid value of not more than 2.5 milligrams potassium hydroxide per gram of fat, and
(viii) a peroxide value of not more than 16 milliequivalents peroxide oxygen per kilogram of fat; and
(c) may contain
(i) lard stearine or fully hydrogenated lard,
(ii) a Class IV preservative, and
(iii) not more than one per cent of substances resulting from the rendering process, other than fatty acids and fat.
- SOR/78-401, s. 1(F)
- SOR/84-300, s. 25(F)
- SOR/2022-168, s. 40
B.09.014 [S]. Leaf Lard shall be lard that has been rendered at a moderately high temperature from the internal fat of the abdomen of the hog, excluding that adhering to the intestines, and shall have an iodine value (Hanus) of not more than 65.
B.09.015 [S]. Suet
(a) shall be the fat taken from the region of the kidney or loin or caul fat of a beef carcass;
(b) shall have
(i) a relative density of not less than 0.893 and not more than 0.898, calculated with the suet at 40°C and water at 20°C (40°C/water at 20°C),
(ii) a refractive index of not less than 1.448 and not more than 1.460, calculated using the sodium D-line as the light source and with the suet at 40°C (nD40°C),
(iii) a titre of not less than 42.5°C and not more than 47°C,
(iv) a saponification value of not less than 190 and not more than 200, expressed as milligrams of potassium hydroxide per gram of fat,
(v) an iodine value of not less than 32 and not more than 47, calculated using the Wijs test,
(vi) an unsaponifiable matter content of not more than 10 grams per kilogram,
(vii) an acid value of not more than 2.0 milligrams potassium hydroxide per gram of fat, and
(viii) a peroxide value of not more than 10 milliequivalents peroxide oxygen per kilogram of fat; and
(c) where sold in comminuted form, shall contain not more than three per cent cereal and one per cent salt.
- SOR/78-655, s. 2(F)
B.09.016 [S]. Margarine
(a) shall be a plastic or fluid emulsion of water in fats, oil or fats and oil that are not derived from milk and may have been subjected to full hydrogenation;
(b) shall contain
(i) not less than 80% fat, oil or fat and oil calculated as fat,
(ii) not less than 3,300 I.U. of vitamin A per 100 g, and
(iii) 26 μg of vitamin D per 100 g; and
(c) may contain
(i) skim milk powder, buttermilk powder or liquid buttermilk,
(ii) whey solids or modified whey solids,
(iii) protein,
(iv) water,
(v) vitamin E, if added in such an amount that will result in the finished product containing not less than 0.6 I.U. of alpha-tocopherol per gram of linoleic acid present in the margarine,
(vi) a flavouring agent,
(vii) a sweetening agent,
(viii) potassium chloride and sodium chloride,
(ix) the following colouring agents: annatto, ß-apo-8′- carotenal, canthaxanthin, carotene, ethyl ß-apo-8′-carotenoate and turmeric, as set out in Table III to section B.16.100,
(x) the following emulsifying agents: lecithin, mono- and di-glycerides, mono-glycerides and sorbitan tristearate, as set out in Table IV to section B.16.100,
(xi) the following pH adjusting agents: citric acid, lactic acid, potassium bicarbonate, sodium bicarbonate, potassium carbonate, sodium carbonate, sodium citrate, sodium lactate, potassium citrate, potassium hydroxide, sodium hydroxide, potassium lactate, sodium potassium tartrate and tartaric acid, as set out in Table X to section B.16.100,
(xii) the following Class II and Class IV preservatives: ascorbyl palmitate, ascorbyl stearate, benzoic acid, butylated hydroxyanisole, butylated hydroxytoluene, calcium sorbate, citric acid esters of mono- and di-glycerides, monoglyceride citrate, monoisopropyl citrate, potassium benzoate, potassium sorbate, propyl gallate, sodium benzoate, sodium sorbate and sorbic acid, as set out in Table XI to section B.16.100, and
(xiii) the following sequestering agents: calcium disodium ethylenediaminetetraacetate and stearyl citrate, as set out in Table XII to section B.16.100.
- SOR/81-60, s. 6
- SOR/84-300, s. 26(F)
- SOR/93-466, s. 1
- SOR/2011-235, s. 1
- SOR/2022-168, s. 41
B.09.017 [S]. Calorie-Reduced Margarine
(a) shall conform to the standard for margarine except it shall contain not more than
(i) 40 per cent fat, oil or fat and oil calculated as fat, and
(ii) 50 per cent of the calories that would be normally present in the product if it were not calorie-reduced;
(b) subject to paragraph (c), may contain, either singly or in combination, in an amount not exceeding 0.5 per cent,
(i) acacia gum,
(ii) agar,
(iii) algin,
(iv) carob bean gum,
(v) carrageenan,
(vi) furcelleran,
(vii) gellan gum,
(viii) guar gum,
(ix) karaya gum,
(x) propylene glycol alginate,
(xi) tragacanth gum, and
(xii) xanthan gum;
(c) may
(i) if it contains none of the ingredients mentioned in paragraph (b), contain polyglycerol esters of fatty acids in an amount not exceeding 0.2 per cent, or
(ii) if it contains a combination of one or more of the ingredients mentioned in paragraph (b) and polyglycerol esters of fatty acids, contain such esters in an amount not exceeding 0.2 per cent, provided that the total combination of such esters and ingredients does not exceed an amount of 0.5 per cent;
(d) notwithstanding subparagraph B.09.016(c)(x), may contain lecithin in an amount not exceeding 0.5 per cent; and
(e) may contain
(i) vegetable starch,
(ii) modified vegetable starch, and
(iii) maltodextrin.
- SOR/94-38, s. 1
- SOR/95-350, s. 1
- SOR/96-160, s. 1
B.09.020 and B.09.021 [Repealed, SOR/88-559, s. 20]
B.09.022 No person shall sell cooking oil, margarine, salad oil, simulated dairy product, shortening or food that resembles margarine or shortening, if the product contains more than five per cent C22 Monoenoic Fatty Acids calculated as a proportion of the total fatty acids contained in the product.
DIVISION 10Flavouring Preparations
B.10.003 [S]. (naming the flavour) Extract or (naming the flavour) Essence shall be a solution in ethyl alcohol, glycerol, propylene glycol or any combination of these, of sapid or odorous principles, or both, derived from the plant after which the flavouring extract or essence is named, and may contain water, a sweetening agent, food colour and a Class II preservative or Class IV preservative.
B.10.004 [S]. Artificial (naming the flavour) Extract, Artificial (naming the flavour) Essence, Imitation (naming the flavour) Extract or Imitation (naming the flavour) Essence shall be a flavouring extract or essence except that the flavouring principles shall be derived in whole, or in part, from sources other than the aromatic plant after which it is named, and if such extract or essence is defined in these Regulations, the flavouring strength of the artificial or imitation extract or essence shall be not less than that of the extract or essence.
B.10.005 [S]. (naming the flavour) Flavour
(a) shall be a preparation, other than a flavouring preparation described in section B.10.003, of sapid or odorous principles, or both, derived from the aromatic plant after which the flavour is named;
(b) may contain a sweetening agent, food colour, Class II preservative, thaumatin, Class IV preservative or emulsifying agent; and
(c) may have added to it the following liquids only:
(i) water,
(ii) any of, or any combination of, the following: benzyl alcohol; 1, 3-butylene glycol, ethyl acetate, ethyl alcohol, glycerol, glyceryl diacetate, glyceryl triacetate, glyceryl tributyrate, isopropyl alcohol, monoglycerides and diglycerides; 1, 2-propylene glycol or triethylcitrate,
(iii) edible vegetable oil, and
(iv) brominated vegetable oil, sucrose acetate isobutyrate or mixtures thereof, when such flavour is used in beverages containing citrus or spruce oils.
- SOR/84-300, s. 27(E)
- SOR/86-1112, s. 1
- SOR/2010-142, s. 8
B.10.006 [S]. Artificial (naming the flavour) Flavour or Imitation (naming the flavour) Flavour shall be a flavour except that the flavouring principles may be derived in whole or in part from sources other than the aromatic plant after which it is named, and if such flavour is defined in these Regulations, the flavouring strength of the artificial or imitation flavour shall be not less than that of the flavour.
B.10.007 [S]. Notwithstanding sections B.10.003 and B.10.005, a (naming the fruit) Extract Naturally Fortified, (naming the fruit) Essence Naturally Fortified or (naming the fruit) Flavour Naturally Fortified shall be an extract, essence or flavour derived from the named fruit to which other natural extractives have been added and 51 per cent of the flavouring strength shall be derived from the named fruit.
B.10.008 On any label of or in any advertisement for an artificial or imitation flavouring preparation, the word “artificial”, or “imitation” shall be an integral part of the name of such flavouring preparation and shall be set out in identical type and identically displayed with such name.
- SOR/84-300, s. 28
B.10.009 [S]. Almond Essence, Almond Extract or Almond Flavour shall be the essence, extract or flavour derived from the kernels of the bitter almond, apricot or peach and shall contain not less than one per cent by volume of hydrocyanic acid-free volatile oil obtained therefrom.
B.10.010 [S]. Anise Essence, Anise Extract or Anise Flavour shall be the essence, extract or flavour derived from natural or terpeneless oil of anise and shall correspond in flavouring strength to an alcoholic solution containing not less than three per cent by volume of oil of anise, the volatile oil obtained from the fruit of Pimpinella anisum L. or Illicium verum Hook.
B.10.011 [S]. Celery Seed Essence, Celery Seed Extract or Celery Seed Flavour shall be the essence, extract or flavour derived from celery seed, or oil of celery seed, or terpeneless oil of celery seed and shall correspond in flavouring strength to an alcoholic solution containing not less than 0.3 per cent by volume of volatile oil of celery seed.
B.10.012 [S]. Cassia Essence, Cassia Extract, Cassia Cinnamon Essence, Cassia Cinnamon Extract, Cassia Flavour or Cassia Cinnamon Flavour shall be the essence, extract or flavour derived from natural or terpeneless oil, obtained from leaves and twigs of Cinnamomum cassia L. containing not less than 80 per cent cinnamic aldehyde, and shall correspond in flavouring strength to an alcoholic solution containing not less than two per cent by volume of volatile oil of cassia cinnamon.
B.10.013 [S]. Ceylon Cinnamon Essence, Ceylon Cinnamon Extract or Ceylon Cinnamon Flavour shall be the essence, extract or flavour derived from the volatile oil obtained from the bark of Cinnamomum zeylanicum Nees, and shall contain
(a) not less than two per cent by volume of oil of Ceylon cinnamon;
(b) not less than 65 per cent cinnamic aldehyde; and
(c) not more than 10 per cent eugenol.
B.10.014 [S]. Clove Essence, Clove Extract or Clove Flavour shall be the essence, extract or flavour derived from the volatile oil obtained from clove buds, and shall contain not less than two per cent by volume of oil of clove.
B.10.015 [S]. Ginger Essence, Ginger Extract or Ginger Flavour shall be the essence, extract or flavour derived from ginger and shall contain in 100 millilitres the alcohol-soluble matter from not less than 20 grams of ginger.
B.10.016 [S]. Lemon Essence, Lemon Extract or Lemon Flavour shall be the essence, extract or flavour prepared from natural or terpeneless oil of lemon or from lemon peel and shall contain not less than 0.2 per cent citral derived from oil of lemon.
B.10.017 [S]. Nutmeg Essence, Nutmeg Extract or Nutmeg Flavour shall be the essence, extract or flavour prepared from natural or terpeneless oil of nutmeg and shall correspond in flavouring strength to an alcoholic solution containing not less than two per cent by volume of oil of nutmeg.
B.10.018 [S]. Orange Essence, Orange Extract or Orange Flavour shall be the essence, extract or flavour prepared from sweet orange peel, oil of sweet orange or terpeneless oil of sweet orange, and shall correspond in flavouring strength to an alcoholic solution containing five per cent by volume of oil of sweet orange, the volatile oil obtained from the fresh peel of Citrus aurantium L., that shall have an optical rotation, at a temperature of 25°C, of not less than +95° using a tube 100 millimetres in length.
B.10.019 [S]. Peppermint Essence, Peppermint Extract or Peppermint Flavour shall be the essence, extract or flavour prepared from peppermint or oil of peppermint, obtained from the leaves and flowering tops of Mentha piperita L., or of Mentha arvensis De.C., var. piperascens Holmes, and shall correspond in flavouring strength to an alcoholic solution of not less than three per cent by volume of oil of peppermint, containing not less than 50 per cent free and combined menthol.
B.10.020 [S]. Rose Essence, Rose Extract or Rose Flavour shall be the essence, extract or flavour prepared from the volatile oil obtained from the petals of Rosa damascena Mill., R. centajolia L., or R. moschata Herrm, and shall contain not less than 0.4 per cent by volume of attar of rose.
B.10.021 [S]. Savory Essence, Savory Extract or Savory Flavour shall be the essence, extract or flavour prepared from savory or oil of savory and shall contain not less than 0.35 per cent by volume oil of savory.
B.10.022 [S]. Spearmint Essence, Spearmint Extract or Spearmint Flavour shall be the essence, extract or flavour prepared from spearmint or from oil of spearmint, obtained from the leaves and flowering tops of Mentha spicata L. and shall contain not less than three per cent by volume of oil of spearmint.
B.10.023 [S]. Sweet Basil Essence, Sweet Basil Extract or Sweet Basil Flavour shall be the essence, extract or flavour prepared from sweet basil or from oil of sweet basil, obtained from the leaves and tops of Ocymum basilicum L. and shall contain not less than 0.1 per cent by volume of oil of sweet basil.
B.10.024 [S]. Sweet Marjoram Essence, Sweet Marjoram Extract, Marjoram Essence, Marjoram Extract, Sweet Marjoram Flavour or Marjoram Flavour shall be the essence, extract or flavour prepared from marjoram or from oil of marjoram and shall contain not less than one per cent by volume of oil of marjoram.
B.10.025 [S]. Thyme Essence, Thyme Extract or Thyme Flavour shall be the essence, extract or flavour prepared from thyme or from oil of thyme and shall contain not less than 0.2 per cent by volume of oil of thyme.
B.10.026 [S]. Vanilla Extract, Vanilla Essence or Vanilla Flavour
(a) shall be the essence, extract or flavour prepared from the vanilla bean, the dried, cured fruit of Vanilla planifolia, Andrews, or Vanilla tahitensia, J. W. Moore;
(b) shall contain in 100 ml, regardless of the method of extraction, at least the quantity of soluble substances in their natural proportions that are extractable, according to official method FO-17, Extraction of Soluble Substances from Vanilla Beans, dated September 15, 1989, from
(i) not less than 10 g of vanilla beans, where the beans contain 25 per cent or less moisture, and
(ii) not less than 7.5 g of vanilla beans on the moisture-free basis, where the beans contain more than 25 per cent moisture; and
(c) notwithstanding sections B.10.003 and B.10.005, shall not contain added colour.
- SOR/82-768, s. 25
- SOR/84-300, s. 29(F)
- SOR/91-149, s. 1
B.10.027 [S]. Wintergreen Essence, Wintergreen Extract or Wintergreen Flavour shall be the essence, extract or flavour prepared from oil of wintergreen, the volatile oil distilled from the leaves of Gaultheria procumbens L. or from Betula lenta L. and shall contain not less than three per cent by volume of oil of wintergreen.
DIVISION 11Fruits, Vegetables, Their Products and Substitutes
- SOR/78-478, s. 1
B.11.001 In this Division,
- acid ingredient
acid ingredient means
(a) citric, malic or tartaric acid,
(b) lemon or lime juice, or
(c) vinegar; (ingrédient acide)
- fruit juice
fruit juice means the unfermented liquid expressed from sound ripe fresh fruit, and includes any such liquid that is heat treated and chilled; (jus de fruit)
- sweetening ingredient
sweetening ingredient means sugar, invert sugar, honey, dextrose, glucose or glucose solids or any combination thereof in dry or liquid form. (ingrédient édulcorant)
B.11.001.1 No person shall sell any fresh fruit or vegetable that is intended to be consumed raw, except grapes, if sulphurous acid or any salt thereof has been added thereto.
- SOR/87-374, s. 1
Vegetables
B.11.002 [S]. Canned (naming the vegetable)
(a) shall be the product obtained by heat processing the named fresh vegetable after it has been properly prepared;
(b) shall be packed in hermetically sealed containers;
(c) may contain
(i) a sweetening ingredient,
(ii) salt,
(iii) water,
(iv) a firming agent, and
(v) citric acid; and
(d) may contain
(i) in the case of canned green beans and canned wax beans, pieces of green peppers, red peppers and tomato in an amount not exceeding 15 per cent of the final product, and dill seasonings and vinegar,
(ii) in the case of canned peas, garnishes composed of one or more of lettuce, onions, carrots, and pieces of green or red peppers in an amount not exceeding 15 per cent of the total drained vegetable ingredient, aromatic herbs, spices and seasonings, stock or juice of vegetables and aromatic herbs, calcium hydroxide in an amount not exceeding 0.01 per cent of the final product and magnesium hydroxide in an amount not exceeding 0.05 per cent of the final product,
(iii) in the case of
(A) canned asparagus, acetic acid, malic acid and tartaric acid in an amount consistent with good manufacturing practice, and
(B) canned white asparagus, acetic acid, ascorbic acid, malic acid and tartaric acid in an amount consistent with good manufacturing practice,
(C) [Repealed, SOR/2012-43, s. 4]
(iv) in the case of asparagus packed in glass containers or fully lined (lacquered) cans, stannous chloride in an amount not exceeding 25 parts per million, calculated as tin,
(v) in the case of canned legumes, other than canned green beans, canned peas and canned wax beans, calcium disodium ethylenediaminetetraacetate in an amount not exceeding 365 parts per million calculated as the anhydrous form, or disodium ethylenediaminetetraacetate in an amount not exceeding 165 parts per million, and
(vi) to (vii) [Repealed, SOR/2012-43, s. 4]
(viii) in the case of canned asparagus, canned green beans, canned wax beans and canned peas
(A) butter or other edible animal or vegetable fats or oils, but if butter is added it shall be not less than three per cent of the final product,
(B) natural or enzymatically or physically modified starches when used with butter or other edible animal or vegetable fats and oils,
(C) acacia gum, algin, carrageenan, furcelleran, guar gum and propylene glycol alginate used with butter or other edible animal or vegetable fats or oils in an amount not exceeding one per cent, singly or in any combination, of the final product, and
(D) characterizing sauces, seasonings or flavouring agents if it is included in the common name of the product.
- SOR/79-660, s. 1
- SOR/84-300, s. 30
- SOR/95-435, s. 1
- SOR/97-561, s. 1
- SOR/2012-43, s. 4
B.11.003 [S]. Canned Mushrooms
(a) shall be the product obtained by heat-processing properly prepared mushrooms of the cultivated type;
(b) shall be packed in hermetically sealed containers; and
(c) may contain ascorbic acid, citric acid and salt.
- SOR/84-300, s. 31
B.11.003A [S]. Frozen Mushrooms
(a) shall be the product obtained by freezing properly prepared mushrooms of the cultivated type; and
(b) may contain citric acid, sodium metabisulphite, sodium phosphate, dibasic, sodium sulphate and salt.
- SOR/2012-43, s. 5
B.11.004 [S]. Frozen (naming the vegetable) shall be the product obtained by freezing the named fresh vegetable after it has been prepared and subjected to a blanching treatment and may contain citric acid and added salt.
- SOR/2012-43, s. 6
B.11.005 [S]. Tomatoes or Canned Tomatoes
(a) shall be the product made by heat processing properly prepared fresh ripe tomatoes;
(b) may contain
(i) a sweetening ingredient in dry form,
(ii) salt,
(iii) a firming agent,
(iv) citric acid, and
(v) spice or other seasoning; and
(c) shall contain not less than 50 per cent drained tomato solids, as determined by official method FO-18, Determination of Drained Tomato Solids, October 15, 1981.
- SOR/82-768, s. 26
B.11.007 [S]. Tomato Juice shall be the unconcentrated, pasteurized liquid containing a substantial portion of fine tomato pulp extracted from sound, ripe, whole tomatoes from which all stems and other portions unfit for consumption have been removed by any method that does not add water to the liquid and may contain citric acid, salt and a sweetening ingredient in dry form.
- SOR/2012-43, s. 7
B.11.009 [S]. Tomato Paste shall be the product made by evaporating a portion of the water from tomatoes or sound tomato trimmings, may contain citric acid, salt and a Class II preservative and shall contain not less than 20 per cent tomato solids as determined by official method FO-19, Determination of Tomato Solids, October 15, 1981.
- SOR/82-768, s. 27
- SOR/2012-43, s. 8
B.11.010 [S]. Concentrated Tomato Paste shall be tomato paste containing not less than 30 per cent tomato solids, as determined by official method FO-19, Determination of Tomato Solids, October 15, 1981.
- SOR/82-768, s. 27
B.11.011 [S]. Tomato Pulp shall be the heat-processed product made from sound, whole and ripe tomatoes or sound tomato trimmings, concentrated to yield a product with a specific gravity of not less than 1.050 (20°C/20°C) and may contain citric acid, salt and a Class II preservative.
- SOR/2012-43, s. 9
B.11.012 [S]. Tomato Puree shall be the heat-processed product made from sound, whole and ripe tomatoes with the skins and seeds removed, concentrated to yield a product with a specific gravity of not less than 1.050 (20°C/20°C) and may contain citric acid, salt and a Class II preservative.
- SOR/2012-43, s. 10
B.11.014 [S]. Tomato Catsup, Catsup or products whose common names are variants of the word Catsup
(a) shall be the heat processed product made from the juice of red-ripe tomatoes or sound tomato trimmings from which skins and seeds have been removed;
(b) shall contain
(i) vinegar,
(ii) salt,
(iii) seasoning, and
(iv) a sweetening ingredient; and
(c) may contain
(i) a Class II preservative, and
(ii) food colour.
B.11.015 [Repealed, SOR/97-151, s. 19]
B.11.016 No person shall sell canned tomatoes, tomato juice or vegetable juice that contains mould filaments in more than 25 per cent of the microscopic fields, when examined by official method MFO-5, Examination of Canned Tomatoes, Tomato Juice and Vegetable Juice, Tomato Puree, Tomato Paste, Tomato Pulp and Tomato Catsup for Mould Filaments, November 30, 1981.
- SOR/82-768, s. 28
B.11.017 No person shall sell tomato puree, tomato paste, tomato pulp or tomato catsup that contains mould filaments in more than 50 per cent of the microscopic fields, when examined by official method MFO-5, Examination of Canned Tomatoes, Tomato Juice and Vegetable Juice, Tomato Puree, Tomato Paste, Tomato Pulp and Tomato Catsup for Mould Filaments, November 30, 1981.
- SOR/82-768, s. 28
B.11.025 No person shall sell potatoes, sweet potatoes or yams that have been artificially coloured.
B.11.040 [S]. Beans with Pork or Beans and Pork shall be the food prepared from dried beans and pork, may contain calcium disodium ethylenediaminetetraacetate, citric acid, disodium ethylenediaminetetraacetate, sauce, seasoning, spices or a sweetening agent and shall contain not less than 60 per cent drained solids as determined by official method FO-20, Determination of Drained Solids of Beans with Pork or Beans and Pork and Beans or Vegetarian Beans, October 15, 1981.
- SOR/82-768, s. 29
- SOR/2012-43, s. 11
B.11.041 [S]. Beans or Vegetarian Beans shall be the food prepared from dried beans, may contain calcium disodium ethylenediaminetetraacetate, citric acid, disodium ethylenediaminetetraacetate, sauce, seasoning, spices or a sweetening agent and shall contain not less than 60 per cent drained solids as determined by official method FO-20, Determination of Drained Solids of Beans with Pork or Beans and Pork and Beans or Vegetarian Beans, October 15, 1981.
- SOR/82-768, s. 29
- SOR/2012-43, s. 12
B.11.050 [S]. Olives shall be the plain or stuffed fruit of the olive tree, and may contain
(a) vinegar;
(b) salt;
(c) a sweetening ingredient;
(d) spices;
(e) seasonings;
(f) lactic acid;
(g) sorbic acid or its potassium or sodium salt;
(h) calcium chloride;
(i) citric acid; and
(j) in the case of ripe olives, ferrous gluconate.
- SOR/97-561, s. 2
B.11.051 [S]. Pickles and relishes shall be the product prepared from vegetables or fruits with salt and vinegar, and may contain
(a) spices;
(b) seasonings;
(c) sugar, invert sugar, dextrose or glucose, in dry or liquid form;
(d) food colour;
(e) a Class II preservative;
(f) a firming agent;
(g) polyoxyethylene (20) sorbitan monooleate in an amount not exceeding 0.05 per cent;
(h) lactic acid;
(h.1) citric acid;
(h.2) sucralose;
(i) vegetable oils; and
(j) in the case of relishes and mustard pickles, a thickening agent.
- SOR/84-300, s. 32
- SOR/2012-43, s. 13
- SOR/2012-104, s. 1
Fruits
B.11.101 [S]. Canned (naming the fruit)
(a) shall be the product prepared by heat processing the named fresh fruit after it has been properly prepared;
(b) shall be packed in hermetically sealed containers;
(c) may contain
(i) a sweetening ingredient,
(ii) water,
(iii) concentrated (naming the fruit) juice, (naming the fruit) juice, (naming the fruits) juice, (naming the fruit) juice from concentrate or any combination thereof,
(iv) citric acid,
(v) acesulfame-potassium, and
(vi) sucralose; and
(d) may contain
(i) in the case of canned pears, lactic acid, lemon juice, malic acid, mint, spice oils, spices, tartaric acid and a flavouring preparation other than that which simulates the flavour of canned pears,
(ii) in the case of canned apples, a firming agent,
(iii) in the case of canned applesauce, ascorbic acid, isoascorbic acid, malic acid, salt, spices and a flavouring preparation other than that which simulates the flavour of canned applesauce,
(iv) in the case of canned grapefruit, calcium chloride, calcium lactate, lemon juice, spices and a flavouring preparation other than that which simulates the flavour of canned grapefruit,
(v) in the case of canned mandarin oranges, ascorbic acid,
(vi) in the case of canned peaches, peach kernels, peach pits and spices intended for flavour development, ascorbic acid and a flavouring preparation other than that which simulates the flavour of canned peaches,
(vii) in the case of canned pineapple, dimethylpolysiloxane when pineapple juice is used as a packing medium, mint, spice oils, spices and a flavouring preparation other than that which simulates the flavour of canned pineapple,
(viii) in the case of canned plums, a flavouring preparation other than that which simulates the flavour of canned plums,
(ix) in the case of canned strawberries, lactic acid, malic acid or tartaric acid, and
(x) in the case of canned apricots, calcium chloride.
- SOR/84-300, s. 33
- SOR/2012-43, s. 14
- SOR/2012-104, s. 2
B.11.102 [S]. Frozen (naming the fruit) shall be the product obtained by freezing the named fresh fruit after it has been properly prepared and may contain
(a) a sweetening ingredient;
(b) water;
(c) fruit juice, fruit juice from concentrate, concentrated fruit juice or any combination thereof;
(d) ascorbic acid, citric acid, erythorbic acid or malic acid to prevent discolouration; and
(e) in the case of frozen sliced apples,
(i) a firming agent, and
(ii) sulphurous acid.
- SOR/84-300, s. 34
- SOR/95-436, s. 1
B.11.103 and B.11.104 [Repealed, SOR/79-252, s. 1]
B.11.105 [Repealed, SOR/97-151, s. 20]
Fruit Juices
B.11.120 [S]. (Naming the fruit) Juice
(a) shall be the juice obtained from the named fruit; and
(b) may contain a sweetening ingredient in dry form, a Class II preservative, amylase, cellulase and pectinase.
- SOR/78-402, s. 2
- SOR/84-300, s. 35
- SOR/90-87, s. 1
- SOR/92-591, s. 2
B.11.121 Notwithstanding section B.11.120, the fruit juice prepared from any fruit named in any of sections B.11.123 to B.11.128A shall conform to the standard prescribed for that fruit juice in that section.
B.11.123 [S]. Apple Juice
(a) shall be the fruit juice obtained from apples;
(b) may contain a Class II preservative, vitamin C, amylase, cellulase and pectinase;
(c) shall have a specific gravity of not less than 1.041 and not more than 1.065 (20°C/20°C); and
(d) shall contain, in 100 millilitres measured at a temperature of 20°C, not less than 0.24 gram and not more than 0.60 gram of ash of which not less than 50 per cent shall be potassium carbonate.
- SOR/90-87, s. 2
B.11.124 [S]. Grape Juice
(a) shall be the fruit juice obtained from grapes;
(b) shall have a specific gravity of not less than 1.040 and not more than 1.124 (20°C/20°C);
(c) shall contain, before the addition of a sweetening ingredient, in 100 millilitres measured at a temperature of 20°C,
(i) not less than 0.20 gram and not more than 0.55 gram of ash, and
(ii) not less than 0.015 gram and not more than 0.070 gram of phosphoric acid calculated as phosphorous pentoxide; and
(d) may contain a pH-adjusting agent, a sweetening ingredient in dry form, a Class II preservative, vitamin C, amylase, cellulase and pectinase.
- SOR/84-300, s. 36(E)
- SOR/86-1112, s. 2
- SOR/90-87, s. 3
B.11.125 [S]. Grapefruit Juice
(a) shall be fruit juice obtained from clean, sound, mature grapefruit;
(b) shall
(i) contain not less than 1.15 milliequivalents of free amino acid per 100 millilitres, as determined by official method FO-21, Determination of Amino Acids in Grapefruit Juice and Orange Juice, October 15, 1981,
(ii) contain not less than 70 milligrams of potassium per 100 millilitres, as determined by official method FO-22, Determination of Potassium in Grapefruit Juice and Orange Juice, October 15, 1981, and
(iii) have an absorbance value for total polyphenolics of not less than 0.310, as determined by official method FO-23, Determination of Absorbance Value for Total Polyphenolics in Grapefruit Juice and Orange Juice, October 15, 1981;
(c) shall, before the addition of sugar, invert sugar, dextrose or glucose solids,
(i) have a Brix reading of not less than 9.3°, as determined by official method FO-24, Determination of Brix Reading for Grapefruit Juice and Orange Juice, October 15, 1981, and
(ii) contain not less than 0.7 per cent and not more than 2.1 per cent of acid by weight calculated as anhydrous citric acid, as determined by official method FO-25, Determination of Acid in Grapefruit Juice or Orange Juice, October 15, 1981; and
(d) may contain sugar, invert sugar, dextrose in dry form, glucose solids, a Class II preservative, amylase, cellulase and pectinase.
- SOR/82-768, s. 30
- SOR/90-87, s. 4
B.11.126 [S]. Lemon Juice
(a) shall be the fruit juice obtained from lemons;
(b) shall contain, before the addition of a sweetening ingredient, in 100 millilitres measured at a temperature of 20°C, not less than
(i) 8.0 grams of soluble solids, as determined by official method FO-26, Determination of Soluble Solids in Lemon Juice, Lime Juice or Lime Fruit Juice, October 15, 1981, and
(ii) 5.0 grams of acid calculated as anhydrous citric acid, as determined by official method FO-27, Determination of Acid in Lemon Juice, Lime Juice, or Lime Fruit Juice, October 15, 1981;
(c) may contain stannous chloride; and
(d) may contain a sweetening ingredient in a dry form, a Class II preservative, amylase, cellulase and pectinase.
- SOR/82-768, s. 31
- SOR/90-87, s. 5
B.11.127 [S]. Lime Juice or Lime Fruit Juice
(a) shall be the fruit juice obtained from limes;
(b) shall have
(i) a specific gravity of not less than 1.030 and not more than 1.040 (20°C/20°C),
(ii) its optical rotation between +0.5 and -1.5 degrees Ventzke, determined at a temperature of 20°C, using a tube 200 millimetres in length;
(c) shall contain, before the addition of a sweetening ingredient, in 100 millilitres measured at a temperature of 20°C, not less than
(i) 8.0 grams of soluble solids, as determined by official method FO-26, Determination of Soluble Solids in Lemon Juice, Lime Juice or Lime Fruit Juice, October 15, 1981, and
(ii) 5.5 grams of acid calculated as anhydrous citric acid, as determined by official method FO-27, Determination of Acid in Lemon Juice, Lime Juice or Lime Fruit Juice, October 15, 1981;
(d) may contain stannous chloride; and
(e) may contain a sweetening ingredient in a dry form, a Class II preservative, amylase, cellulase and pectinase.
- SOR/82-768, s. 32
- SOR/90-87, s. 6
B.11.128 [S]. Orange Juice
(a) shall be fruit juice obtained from clean, sound, mature oranges;
(b) shall
(i) contain not less than 1.20 milliequivalents of free amino acids per 100 millilitres, as determined by official method FO-21, Determination of Amino Acids in Grapefruit Juice and Orange Juice, October 15, 1981,
(ii) contain not less than 115 milligrams of potassium per 100 millilitres, as determined by official method FO-22, Determination of Potassium in Grapefruit Juice and Orange Juice, October 15, 1981, and
(iii) have an absorbance value for total polyphenolics of not less than 0.380, as determined by official method FO-23, Determination of Absorbance Value for Total Polyphenolics in Grapefruit Juice and Orange Juice, October 15, 1981;
(c) shall, before the addition of sugar, invert sugar, dextrose or glucose solids,
(i) have a Brix reading of not less than 9.7°, as determined by official method FO-24, Determination of Brix Reading for Grapefruit Juice and Orange Juice, October 15, 1981, and
(ii) contain not less than 0.5 per cent and not more than 1.8 per cent of acid by weight calculated as anhydrous citric acid, as determined by official method FO-25, Determination of Acid in Grapefruit Juice or Orange Juice, October 15, 1981;
(d) may contain orange essences, orange oils and orange pulp adjusted in accordance with good manufacturing practice; and
(e) may contain sugar, invert sugar, dextrose in dry form, glucose solids, a Class II preservative, amylase, cellulase and pectinase.
- SOR/82-768, s. 33
- SOR/90-87, s. 7
B.11.128A [S]. Pineapple Juice
(a) shall be the fruit juice obtained from pineapple; and
(b) may contain a sweetening ingredient in dry form, a Class II preservative, vitamin C, amylase, cellulase, pectinase and an antifoaming agent.
- SOR/90-87, s. 8
- SOR/91-90, s. 1
B.11.129 [S]. Carbonated (naming the fruit) Juice or Sparkling (naming the fruit) Juice shall be the named fruit juice impregnated with carbon dioxide under pressure.
B.11.130 [S]. (1) Concentrated (naming the fruit) juice
(a) shall be fruit juice that is concentrated to at least one half of its original volume by the removal of water;
(b) may contain
(i) vitamin C,
(ii) food colour,
(iii) stannous chloride,
(iv) a sweetening ingredient, and
(v) a class II preservative; and
(c) may have added to it, for the purpose of adjustment in accordance with good manufacturing practice, all or any of the following, namely,
(i) essence, oil and pulp from the named fruit, and
(ii) water.
(2) Subparagraphs (1)(b)(i), (ii), (iii) and (v) do not apply in respect of frozen concentrated orange juice.
- SOR/89-198, s. 2
- SOR/91-124, s. 3
B.11.131 [S]. (Naming the fruits) Juice shall be a mixture of fruit juices each of which meets the standard prescribed for that fruit juice in this Division.
B.11.132 [S]. Apple and (naming the fruit) Juice
(a) shall be a mixture of apple juice and another fruit juice, each of which meets the standard, if any, prescribed for that fruit juice in this Division; and
(b) may contain added vitamin C.
B.11.133 [S]. Reconstituted (naming the fruit) Juice or (naming the fruit) Juice from Concentrate
(a) shall be fruit juice that has been prepared by the addition of water to fruit juice of the same name from which water has been removed;
(b) may contain juice of the same name, a sweetening ingredient, and natural pulp, oils and esters of the named fruit;
(c) shall conform to the standards for the named fruit juices as prescribed in this Division; and
(d) may contain, in the case of reconstituted lemon or lime juice, not more than 10 parts per million dimethylpolysiloxane.
- SOR/78-637, s. 2
B.11.134 [S]. Apricot Nectar, Peach Nectar or Pear Nectar
(a) shall be the unfermented but fermentable pulpy product, intended for direct consumption, obtained by blending the total edible part of sound and ripe apricots, peaches and pears, as the case may be, concentrated or unconcentrated with water and, subject to subparagraph (e)(i), a sweetening ingredient;
(b) shall contain
(i) in the case of peach nectar and pear nectar, not less than 40 per cent by weight of the fruit or the equivalent derived from the concentrated fruit, and
(ii) in the case of apricot nectar, not less than 35 per cent by weight of the fruit or the equivalent derived from the concentrated fruit;
(c) shall contain not less than 13 per cent soluble solids by weight expressed as °Brix on the International Sucrose Scales and calculated by refractometer at 20°C and uncorrected for acidity;
(d) shall not contain more than 3 g/kg (3000 p.p.m.) of ethanol and 10 mg/kg (10 p.p.m.) of hydroxy methyl furfural; and
(e) may contain
(i) honey if no other sweetening ingredient is employed,
(ii) citric acid and malic acid at levels consistent with good manufacturing practice,
(iii) lemon juice, and
(iv) vitamin C.
- SOR/79-660, s. 2
- SOR/2010-94, s. 9(E)
Fruit Flavoured Drinks
B.11.150 No person shall label, package, sell or advertise a fruit flavoured drink in a manner that is likely to create an impression that the fruit flavoured drink contains vitamins or has any other nutritional value commonly associated with any fruit juice unless the following requirements are met:
(a) it is sold as a substitute for fruit juice or as a breakfast drink;
(b) it is not carbonated;
(c) it is not represented to be or is not commonly known as
(i) a soft drink, or
(ii) a thirst-quenching or refreshment drink; and
(d) notwithstanding sections D.01.009, D.01.011 and D.02.009,
(i) it contains vitamin C in an amount not less than 24 milligrams and not more than 48 milligrams, and
(ii) it contains
(A) where folic acid has been added, an amount of folic acid of not less than 40 micrograms and not more than 80 micrograms,
(B) where thiamine has been added, an amount of thiamine of not less than 0.08 milligram and not more than 0.11 milligram,
(C) where iron has been added, an amount of iron of not less than 0.56 milligram and not more than 0.80 milligram, or
(D) where potassium has been added, an amount of potassium of not less than 100 milligrams and not more than 200 milligrams,
per 100 millilitres when the drink is ready to serve.
- SOR/78-478, s. 2
B.11.151 No person shall label, package, sell or advertise a base, concentrate or mix that is used for making a fruit flavoured drink in a manner that is likely to create an impression that the drink made therefrom will contain vitamins or have any other nutritional value commonly associated with fruit juice unless the following requirements are met:
(a) the base, concentrate or mix
(i) is sold for the purpose of making a breakfast drink or a substitute for fruit juice,
(ii) is not represented to be or is not commonly known as a product that is used for making a soft drink or a thirst-quenching or refreshment drink; and
(b) where a drink is made therefrom as directed, the drink meets the requirements described in paragraph B.11.150(d).
- SOR/78-478, s. 2
Jams
B.11.201 [S]. (Naming the fruit) Jam
(a) shall be the product obtained by processing fruit, fruit pulp, or canned fruit, by boiling to a suitable consistency with water and a sweetening ingredient;
(b) shall contain not less than
(i) 45 per cent of the named fruit, and
(ii) 66 per cent water soluble solids as estimated by the refractometer;
(c) may contain
(i) such amount of added pectin, pectinous preparation, or acid ingredient as reasonably compensates for any deficiency in the natural pectin content or acidity of the named fruit,
(ii) a Class II preservative,
(iii) a pH adjusting agent, and
(iv) an antifoaming agent; and
(d) shall not contain apple or rhubarb.
- SOR/92-400, s. 16
B.11.202 [S]. (Naming the fruit) Jam with Pectin
(a) shall be the product obtained by processing fruit, fruit pulp, or canned fruit by boiling to a suitable consistency with water and a sweetening ingredient;
(b) shall contain
(i) not less than 27 per cent of the named fruit,
(ii) not less than 66 per cent water soluble solids as estimated by the refractometer, and
(iii) pectin or pectinous preparations;
(c) may contain
(i) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the named fruit,
(ii) food colour,
(iii) a Class II preservative,
(iv) a pH adjusting agent, and
(v) an antifoaming agent; and
(d) shall not contain apple or rhubarb.
- SOR/92-400, s. 17
B.11.203 [S]. Apple (or Rhubarb) and (naming the fruit) Jam
(a) shall be the product obtained by processing fruit, fruit pulp or canned fruit by boiling to a suitable consistency with water and a sweetening ingredient;
(b) shall contain not less than
(i) 12.5 per cent of the named fruit, except that where the named fruit is strawberry it shall contain not less than 15 per cent strawberries,
(ii) 20 per cent apple or rhubarb pulp, and
(iii) 66 per cent water soluble solids as estimated by the refractometer; and
(c) may contain
(i) pectin or pectinous preparation,
(ii) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the fruit used in its preparation,
(iii) food colour,
(iv) a Class II preservative,
(v) a pH adjusting agent, and
(vi) an antifoaming agent.
B.11.204 [Repealed, SOR/2022-143, s. 21]
Marmalade
B.11.220 [S]. (Naming the citrus fruit) Marmalade shall be the food of jelly-like consistency made from any combination of peel, pulp or juice of the named citrus fruit by boiling with water and a sweetening ingredient and shall contain not less than 65 per cent water soluble solids as estimated by the refractometer and may contain
(a) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the named citrus fruit;
(b) a pH adjusting agent; and
(c) an antifoaming agent.
B.11.221 [S]. (Naming the citrus fruit) Marmalade with Pectin
(a) shall be the food of jelly-like consistency made from any combination of peel, pulp or juice of the named citrus fruit by boiling with water and a sweetening ingredient;
(b) shall contain
(i) not less than 27 per cent of any combination of peel, pulp or juice of the named citrus fruit,
(ii) not less than 65 per cent water soluble solids as estimated by the refractometer, and
(iii) pectin or pectinous preparation; and
(c) may contain
(i) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the citrus fruit used in its preparation,
(ii) a Class II preservative,
(iii) a pH adjusting agent, and
(iv) an antifoaming agent.
B.11.222 [S]. Pineapple Marmalade or Fig Marmalade
(a) shall be the food of jelly-like consistency made from the pulp of juice of the named fruit by boiling with water and a sweetening ingredient;
(b) shall contain not less than
(i) 45 per cent of the named fruit, and
(ii) 65 per cent water soluble solids, as estimated by the refractometer;
(c) may contain such amounts of added pectin, pectinous preparation or acid ingredient as reasonably compensates for any deficiency in the natural pectin content or acidity of the named fruit;
(d) a pH adjusting agent; and
(e) an antifoaming agent.
B.11.223 [S]. Pineapple Marmalade with Pectin or Fig Marmalade with Pectin
(a) shall be the food of jelly-like consistency made from the pulp and juice of the named fruit by boiling with water and a sweetening ingredient;
(b) shall contain
(i) not less than 27 per cent of the named fruit,
(ii) not less than 65 per cent water soluble solids as estimated by the refractometer, and
(iii) pectin or pectinous preparation; and
(c) may contain
(i) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the named fruit,
(ii) food colour,
(iii) a Class II preservative,
(iv) a pH adjusting agent, and
(v) an antifoaming agent.
- SOR/84-300, s. 37(E)
B.11.224 [S]. (Naming the fruit) Preserve (Conserve) shall be the food made by processing fruit other than apple or rhubarb with a sweetening ingredient and shall contain not less than
(a) 45 parts by weight of the named fruit for each 55 parts by weight, on the dry basis, of a sweetening ingredient; and
(b) 60 per cent water-soluble solids, as estimated by the refractometer.
Jelly
B.11.240 [S]. (Naming the fruit) Jelly shall be the gelatinous food, free of seeds and pulp, made from the named fruit, the juice of the named fruit or a concentrate of the juice of the named fruit, which has been boiled with water and a sweetening ingredient, shall contain not less than 62 per cent water soluble solids as estimated by the refractometer and may contain
(a) such amount of added pectin, pectinous preparation or acid ingredient as reasonably compensates for any deficiency of the natural pectin content or acidity of the named fruit;
(b) a pH adjusting agent; and
(c) an antifoaming agent.
B.11.241 [S]. (Naming the fruit) Jelly with Pectin
(a) shall be the gelatinous food, free of seeds and pulp, made from the named fruit, the juice of the named fruit or a concentrate of the juice of the named fruit, which has been boiled with water and a sweetening ingredient;
(b) shall contain
(i) not less than the equivalent of 32 per cent juice of the named fruit,
(ii) not less than 62 per cent water soluble solids, as estimated by the refractometer, and
(iii) pectin or pectinous preparation; and
(c) may contain
(i) such amount of acid ingredient as reasonably compensates for any deficiency in the natural acidity of the named fruit,
(ii) juice of another fruit,
(iii) a gelling agent,
(iv) food colour,
(v) a Class II preservative,
(vi) a pH adjusting agent, and
(vii) an antifoaming agent.
B.11.242 The standards prescribed in these Regulations for jam and jelly do not apply to cranberry sauce, jellied cranberry, cranberry jelly, mint jelly and jellied mint.
Mincemeat
B.11.250 [S]. Mincemeat, Mince Meat, Mince or Fruit Mince
(a) shall be the food prepared from
(i) fruit or dried fruit,
(ii) suet,
(iii) salt,
(iv) spices, and
(v) a sweetening agent; and
(b) may contain
(i) vinegar,
(ii) fresh, concentrated or fermented fruit juice,
(iii) spiritous liquor,
(iv) nuts,
(v) cooked meat,
(vi) a Class II preservative,
(vii) a thickening agent,
(viii) citric acid, and
(ix) caramel.
- SOR/84-300, s. 38(F)
- SOR/2011-278, s. 9
Boiled Cider
B.11.260 [S]. Boiled Cider shall be the liquid expressed from whole apples, apple cores, apple trimmings or apple culls and concentrated by boiling.
DIVISION 12Prepackaged Water and Ice
- SOR/80-633, s. 1
B.12.001 [S]. Water represented as mineral water or spring water,
(a) shall be potable water obtained from an underground source but not obtained from a public community water supply;
(b) shall not contain any coliform bacteria, as determined by official method MFO-9, Microbiological Examination of Mineral Water, November 30, 1981;
(c) shall not have its composition modified through the use of any chemicals; and
(d) notwithstanding paragraph (c), may contain
(i) added carbon dioxide,
(ii) added fluoride, if the total fluoride ion content thereof does not exceed one part per million, and
(iii) added ozone.
- SOR/80-633, s. 2
- SOR/82-768, s. 34
B.12.002 The label on a container of water represented as mineral water or spring water shall carry a statement
(a) of the geographical location of the underground source from which it is obtained;
(b) of the total dissolved mineral salt content expressed in parts per million; and
(c) of the total fluoride ion content expressed in parts per million.
(d) [Repealed, SOR/2022-143, s. 22]
- SOR/84-300, s. 39(F)
- SOR/88-336, s. 3
- SOR/92-626, s. 14(F)
- SOR/2022-143, s. 22
B.12.003 [Repealed, SOR/2022-143, s. 23]
B.12.004 No person shall sell water in sealed containers, other than water represented as mineral water or spring water, if it contains
(a) any coliform bacteria, as determined by official method MFO-15, Microbiological Examination of Water in Sealed Containers (Excluding Mineral and Spring Water) and of Prepackaged Ice, November 30, 1981;
(b) more than 100 total aerobic bacteria per millilitre, as determined by official method MFO-15, Microbiological Examination of Water in Sealed Containers (Excluding Mineral and Spring Water) and of Prepackaged Ice, November 30, 1981;
(c) naturally occurring fluoride ion in an amount that exceeds its naturally occurring amount; or
(d) added fluoride in such an amount that the total amount therein of added and naturally occurring fluoride ion exceeds one part per million.
- SOR/80-633, s. 3
- SOR/82-768, s. 35
B.12.005 (1) No person shall sell prepackaged ice if it contains
(a) any coliform bacteria, as determined by official method MFO-15, Microbiological Examination of Water in Sealed Containers (Excluding Mineral and Spring Water) and of Prepackaged Ice, November 30, 1981;
(b) naturally occurring fluoride ion in an amount that exceeds its naturally occurring amount; or
(c) added fluoride in such an amount that the total amount therein of added and naturally occurring fluoride ion exceeds one part per million.
(2) No person shall manufacture prepackaged ice for sale if the water from which it is made contains
(a) any coliform bacteria, as determined by official method MFO-15, Microbiological Examination of Water in Sealed Containers (Excluding Mineral and Spring Water) and of Prepackaged Ice, November 30, 1981;
(b) naturally occurring fluoride ion in an amount that exceeds its naturally occurring amount; or
(c) added fluoride in such an amount that the total amount therein of added and naturally occurring fluoride ion exceeds one part per million.
- SOR/80-633, s. 3
- SOR/82-768, s. 36
B.12.006 [Repealed, SOR/2022-143, s. 24]
B.12.007 Notwithstanding section B.01.008, when chlorine or any compounds of chlorine have been
(a) used in the treatment of water in sealed containers, other than water represented as mineral water or spring water, and
(b) subsequently removed from the water together with any chlorine and compounds of chlorine produced in the water,
chlorine or any compounds of chlorine need not be shown as ingredients on any part of the label on a sealed container of that water.
- SOR/80-633, s. 3
B.12.008 A statement of the total fluoride ion content expressed in parts per million shall appear on the principal display panel of the label on a sealed container of water, other than water represented as mineral water or spring water and on the label on a container of prepackaged ice.
- SOR/80-633, s. 3
- SOR/2000-353, s. 5(E)
B.12.009 [Repealed, SOR/2022-143, s. 25]
DIVISION 13Grain and Bakery Products
B.13.001 [S]. Flour, White Flour, Enriched Flour or Enriched White Flour
(a) shall be the food prepared by the grinding and bolting through cloth having openings not larger than those of woven wire cloth designated “149 microns (No. 100)”, of cleaned milling grades of wheat;
(b) shall be free from bran coat and germ to such an extent that the percentage of ash therein, before the addition of any other material permitted by this section, calculated on a moisture-free basis, does not exceed 1.20 per cent;
(c) shall have a moisture content of not more than 15 per cent;
(d) shall contain in 100 grams of flour
(i) 0.64 milligram of thiamine,
(ii) 0.40 milligram of riboflavin,
(iii) 5.30 milligrams of niacin or niacinamide,
(iv) 0.15 milligram of folic acid, and
(v) 4.4 milligrams of iron;
(e) may contain
(i) malted wheat flour,
(ii) malted barley flour in an amount not exceeding 0.50 per cent of the weight of the flour,
(iii) amylase, amylase (maltogenic), asparaginase, bromelain, glucoamylase, glucose oxidase, lactase, lipase, lipoxidase, pentosanase, phospholipase, protease, pullulanase or xylanase,
(iv) chlorine,
(v) chlorine dioxide,
(vi) benzoyl peroxide in an amount not exceeding 150 parts by weight for each one million parts of flour, with or without not more than 900 parts by weight for each one million parts of flour of one or a mixture of two or more of calcium carbonate, calcium sulphate, dicalcium phosphate, magnesium carbonate, potassium aluminum sulphate, sodium aluminum sulphate, starch and tricalcium phosphate as carriers of the benzoyl peroxide,
(vii) [Repealed, SOR/94-227, s. 1]
(viii) ammonium persulphate in an amount not exceeding 250 parts by weight for each one million parts of flour,
(ix) ammonium chloride in an amount not exceeding 2,000 parts by weight for each one million parts of flour,
(x) acetone peroxide,
(xi) azodicarbonamide in an amount not exceeding 45 parts by weight for each one million parts of flour,
(xii) ascorbic acid in an amount not exceeding 200 parts by weight for each one million parts of flour,
(xiii) l-cysteine (hydrochloride) in an amount not exceeding 90 parts by weight for each one million parts of flour,
(xiv) monocalcium phosphate in an amount not exceeding 7,500 parts by weight for each one million parts of flour, and
(xv) in 100 grams of flour
(A) 0.31 milligram of vitamin B6,
(B) 1.3 milligrams of d-pantothenic acid, and
(C) 190 milligrams of magnesium; and
(f) may contain calcium carbonate, edible bone meal, chalk (B.P.), ground limestone or calcium sulphate in an amount that will provide in 100 grams of flour 140 milligrams of calcium.
(g) [Repealed, SOR/97-151, s. 21]
- SOR/78-402, s. 3
- SOR/78-698, s. 2
- SOR/80-632, s. 3
- SOR/82-383, s. 5
- SOR/84-300, s. 41(E)
- SOR/89-145, s. 1
- SOR/92-63, s. 1
- SOR/92-94, s. 1
- SOR/94-227, s. 1
- SOR/94-689, s. 2
- SOR/96-527, s. 1
- SOR/97-122, s. 1
- SOR/97-151, s. 21
- SOR/97-558, s. 1
- SOR/98-550, s. 1
- SOR/2003-130, s. 1
- SOR/2012-26, s. 1
- SOR/2012-46, s. 1
B.13.002 Notwithstanding section B.13.001, flour, white flour, enriched flour or enriched white flour, used in or sold for the manufacture of gluten or starch is not required to contain added thiamine, riboflavin, niacin, folic acid or iron.
- SOR/98-550, s. 2
B.13.003 [S]. Vitamin B White Flour (Canada Approved)
(a) shall be flour that has been milled in such a way as to retain a high proportion of the vitamins naturally occurring in the original wheat berry;
(b) shall constitute not less than 70 per cent of the wheat from which it is milled;
(c) shall be bolted through at least one cloth having openings not larger than those of woven wire cloth designated “149 microns (No. 100)”; and
(d) shall contain, on a moisture-free basis,
(i) in one pound an amount of the vitamin B complex that will contribute not less than 1.2 milligrams of thiamine, and
(ii) not more than 0.70 per cent and not less than 0.61 per cent ash.
B.13.004 [Repealed, SOR/79-252, s. 2]
B.13.005 [S]. Whole Wheat Flour or Entire Wheat Flour
(a) shall be the food prepared by the grinding and bolting of cleaned, milling grades of wheat from which a part of the outer bran or epidermis layer may have been separated;
(b) shall contain the natural constituents of the wheat berry to the extent of not less than 95 per cent of the total weight of the wheat from which it is milled;
(c) shall have
(i) an ash content, calculated on a moisture-free basis, of not less than 1.25 per cent and not more than 2.25 per cent,
(ii) a moisture content of not more than 15 per cent, and
(iii) such a degree of fineness that not less than 90 per cent bolts freely through a No. 8 (2 380 micron) sieve, and not less than 50 per cent through a No. 20 (840 micron) sieve; and
(d) may contain
(i) malted wheat flour,
(ii) malted barley flour in an amount not exceeding 0.50 per cent of the weight of the flour,
(iii) amylase, amylase (maltogenic), asparaginase, bromelain, glucoamylase, glucose oxidase, lactase, lipase, lipoxidase, pentosanase, phospholipase, protease, pullulanase or xylanase,
(iv) chlorine,
(v) chlorine dioxide,
(vi) benzoyl peroxide in an amount not exceeding 150 parts by weight for each one million parts of flour, with or without not more than 900 parts by weight for each one million parts of flour of one or a mixture of two or more of calcium carbonate, calcium sulphate, dicalcium phosphate, magnesium carbonate, potassium aluminum sulphate, sodium aluminum sulphate, starch and tricalcium phosphate as carriers of the benzoyl peroxide,
(vii) [Repealed, SOR/94-227, s. 2]
(viii) ammonium persulphate in an amount not exceeding 250 parts by weight for each one million parts of flour,
(ix) ammonium chloride in an amount not exceeding 2,000 parts by weight for each one million parts of flour,
(x) azodicarbonamide in an amount not exceeding 45 parts by weight for each one million parts of flour,
(xi) acetone peroxide,
(xii) ascorbic acid in an amount not exceeding 200 parts by weight for each one million parts of flour, and
(xiii) l-cysteine (hydrochloride) in an amount not exceeding 90 parts by weight for each one million parts of flour.
(e) [Repealed, SOR/97-151, s. 22]
- SOR/78-402, s. 4
- SOR/80-632, s. 4
- SOR/82-383, s. 6
- SOR/92-63, s. 2
- SOR/92-94, s. 2
- SOR/94-227, s. 2
- SOR/94-689, s. 2
- SOR/97-122, s. 2
- SOR/97-151, s. 22
- SOR/97-558, s. 2
- SOR/2000-184, s. 63(F)
- SOR/2003-130, s. 2
- SOR/2012-26, s. 2
- SOR/2012-46, s. 2
B.13.006 [S]. Graham Flour shall be flour to which has been added part of the bran and other constituents of the wheat berry, and shall have an ash content, calculated on a moisture-free basis, of not less than 1.20 per cent and not more than 2.25 per cent.
B.13.007 [S]. Gluten Flour shall be the food obtained by removing from flour a part of the starch and shall not contain more than
(a) 10 per cent moisture, and
(b) 44 per cent Starch, calculated on a moisture-free basis, as determined by official method FO-28, Determination of Starch in Gluten Flour, October 15, 1981.
- SOR/82-768, s. 37
B.13.008 [S]. Crushed Wheat or Coarse Ground Wheat shall be the food prepared by so crushing cleaned wheat that 40 per cent or more passes through a No. 8 (2 380 micron) sieve and less than 50 per cent through a No. 20 (840 micron) sieve, the proportions of the natural constituents of such wheat, other than moisture, remaining unaltered and shall have
(a) an ash content, calculated on a moisture-free basis, of not less than 1.50 per cent and not more than 2.25 per cent; and
(b) a moisture content of not more than 15.5 per cent.
B.13.009 [S]. Cracked Wheat shall be the food prepared by so cracking or cutting cleaned wheat into angular fragments that not less than 90 per cent passes through a No. 8 (2 380 micron) sieve and not more than 20 per cent through a No. 20 (840 micron) sieve, the proportions of the natural constituents of such wheat, other than moisture, remaining unaltered and shall have
(a) an ash content, calculated on a moisture-free basis, of not less than 1.50 per cent and not more than 2.25 per cent; and
(b) a moisture content of not more than 15.5 per cent.
B.13.010 [S]. Rice shall be the hulled or hulled and polished seed of the rice plant and, in the case of hulled and polished seeds, may be coated with magnesium silicate, talc and glucose.
- SOR/78-403, s. 3
B.13.010.1 (1) For the purposes of this Division, precooked rice means polished rice that has been cooked in water or steam and dried in such a manner as to retain the rice grains in a porous and open-structured condition.
(2) Notwithstanding sections D.01.009, D.01.011 and D.02.009, no person shall sell pre-cooked rice to which a vitamin or mineral nutrient set out in Column I of an item of the table to this section has been added, either singly or in any combination, unless each 100 g of the pre-cooked rice as sold contains the added vitamin or mineral nutrient in the amount set out in Column II of that item.
TABLE
Column I Column II Item Vitamin or Mineral Nutrient Amount per 100 g of Pre-cooked Rice 1 Thiamine 0.45 mg 2 Niacin 4.2 mg 3 Vitamin B6 0.6 mg 4 Folic acid 0.016 mg 5 Pantothenic acid 1.2 mg 6 Iron 1.6 mg (3) No person shall represent pre-cooked rice as “enriched” unless the food contains added thiamine, niacin and iron.
- SOR/86-320, s. 1
- SOR/98-458, s. 7(F)
B.13.011 [S]. Corn starch shall be starch made from maize and shall contain not less than 84% starch.
- SOR/84-300, s. 42
- SOR/2011-28, s. 5
B.13.014 For the purpose of this Division, moisture, ash and fineness shall be determined by the following applicable official methods:
(a) FO-29, Determination of Moisture in Grain, October 15, 1981;
(b) FO-30, Determination of Ash in Grain, October 15, 1981; and
(c) FO-31, Determination of Degree of Fineness of Grain, October 15, 1981.
- SOR/82-768, s. 38
B.13.015 [S]. Cottonseed Flour or similar products from cottonseed shall be derived from decorticated, defatted or partially defatted, cooked, ground cottonseed kernels and contain not more than 450 parts per million of free gossypol.
B.13.020 In this Division, milk solids means the entire solids content from milk, partly skimmed milk or skim milk or their concentrated, dried or reconstituted form, singly or in any combination.
- SOR/89-170, s. 1
Bread
B.13.021 [S]. Bread or White Bread shall be the food made by baking a yeast-leavened dough prepared with flour and water and may contain
(a) salt;
(b) shortening, lard, butter or margarine;
(c) milk or milk product;
(d) whole egg, egg-white; egg-yolk, (fresh, dried, or frozen);
(e) a sweetening agent;
(f) malt syrup, malt extract or malt flour;
(g) inactive dried yeast of the genus Saccharomyces cerevisiae in an amount not greater than two parts by weight for each 100 parts of flour used;
(h) amylase, amylase (maltogenic), asparaginase, bromelain, glucoamylase, glucose oxidase, lactase, lipase, lipoxidase, pentosanase, phospholipase, protease, pullulanase or xylanase;
(i) subject to section B.13.029, one or more of the following in a total amount not exceeding five parts by weight per 100 parts of flour used, namely, whole wheat flour, entire wheat flour, graham flour, gluten flour, wheat meal, wheat starch, non-wheat flour, non-wheat meal or non-wheat starch, any of which may be wholly or partially dextrinized;
(j) other parts of the wheat berry;
(k) lecithin or ammonium salt of phosphorylated glyceride;
(l) monoglycerides and diglycerides of fat-forming fatty acids,
(m) ammonium chloride, ammonium sulphate, calcium carbonate, calcium lactate, diammonium phosphate, dicalcium phosphate, monoammonium phosphate or any combination thereof in an amount not greater than 0.25 parts by weight of all such additives for each 100 parts of flour used;
(n) monocalcium phosphate in an amount not greater than 0.75 parts by weight for each 100 parts of flour used;
(o) calcium peroxide, ammonium persulphate, potassium persulphate or any combination thereof in an amount not greater than 0.01 part by weight of all such additives for each 100 parts of flour used;
(p) acetone peroxide;
(q) vinegar;
(r) Class III preservative;
(s) food colour;
(t) calcium stearoyl-2-lactylate or sodium stearoyl-2-lactylate in an amount not greater than 0.375 parts by weight for each 100 parts of flour used;
(u) l-cysteine (hydrochloride) in an amount not greater than 0.009 parts by weight for each 100 parts of flour used;
(v) calcium sulphate in an amount not greater than 0.5 parts by weight for each 100 parts of flour used;
(w) sodium stearyl fumarate in an amount not greater than 0.5 parts by weight for each 100 parts of flour used;
(x) ascorbic acid in an amount not greater than 0.02 parts by weight for each 100 parts of flour used;
(y) lactic acid;
(z) azodicarbonamide in an amount not exceeding 45 parts by weight for each one million parts of flour;
(aa) calcium iodate, potassium iodate or any combination thereof in an amount not greater than 45 parts by weight of all such additives for each one million parts of flour;
(bb) acetylated tartaric acid esters of mono- and diglycerides in an amount not greater than 0.6 parts by weight for each 100 parts of flour used; and
(cc) guar gum.
- SOR/78-402, s. 5
- SOR/79-251, s. 2
- SOR/82-383, ss. 7, 8
- SOR/84-300, s. 43(E)
- SOR/92-63, s. 3
- SOR/92-94, s. 3
- SOR/94-227, s. 3
- SOR/97-122, s. 3
- SOR/97-558, s. 3
- SOR/2003-130, s. 3
- SOR/2007-302, s. 4(F)
- SOR/2011-278, s. 10
- SOR/2012-26, s. 3
- SOR/2012-43, s. 15(F)
- SOR/2012-46, s. 3
B.13.022 [S]. Enriched Bread or Enriched White Bread
(a) shall be bread that is baked from a dough in which enriched flour is the only wheat flour used;
(b) shall contain
(i) for each 100 parts of flour used, not less than
(A) two parts by weight of skim milk solids,
(B) four parts by weight of dried whey powder, or
(C) such amount of the protein product made from peas (Pisum sativum) or soybeans (Glycine max) as will provide 0.5 parts by weight of protein, and
(ii) in 100 grams of bread,
(A) 0.40 milligram of thiamine,
(B) 0.24 milligram of riboflavin,
(C) 3.3 milligrams of niacin or niacinamide,
(D) 0.10 milligram of folic acid, and
(E) 2.76 milligrams of iron;
(c) may contain, in 100 grams of bread,
(i) 0.14 milligram of vitamin B6,
(ii) 0.6 milligram of d-pantothenic acid,
(iii) 90 milligrams of magnesium, and
(iv) 66 milligrams of calcium; and
(d) where it contains not less than six parts by weight of milk solids per 100 parts of enriched flour used, may be described by the common name “milk bread”.
- SOR/78-698, s. 3
- SOR/87-704, s. 1
- SOR/89-170, s. 2
- SOR/89-198, s. 3
- SOR/98-550, s. 3
B.13.023 and B.13.024 [Repealed, SOR/79-252, s. 3]
B.13.025 [S]. Raisin Bread shall be bread that contains for each 100 parts by weight of flour used not less than 50 parts by weight of seeded or seedless raisins, or raisins and currants of which not less than 35 parts shall be raisins and may contain spices or peel.
B.13.026 [S]. (naming the percentage) Whole Wheat Bread
(a) shall
(i) be bread in the making of which the named percentage of the flour used shall be whole wheat flour, and
(ii) contain not less than 60 per cent whole wheat flour in relation to the total flour used; and
(b) may
(i) contain caramel, and
(ii) where it contains not less than six parts by weight of milk solids per 100 parts of the total enriched flour and whole wheat flour used, be described by the common name “(naming the percentage) whole wheat milk bread”.
- SOR/89-170, s. 3
B.13.027 [S]. Brown Bread shall be bread coloured by the use of whole wheat flour, graham flour, bran, molasses or caramel.
B.13.028 [Repealed, SOR/97-151, s. 23]
B.13.029 A specialty bread may contain
(a) one or more of the ingredients specified in paragraph B.13.021(i) in a total amount greater than the total amount specified in that paragraph; and
(b) fruit, nuts, seeds and flavouring.
- SOR/79-251, s. 3
Alimentary Paste
B.13.051 No person shall sell macaroni, spaghetti, noodles or similar alimentary pastes, as egg macaroni, egg spaghetti, egg noodles or egg alimentary pastes, respectively, unless they contain, on the dry basis, not less than four per cent, egg-yolk solids derived from whole egg, dried egg, frozen egg or frozen egg-yolk.
B.13.052 (1) Notwithstanding sections D.01.009, D.01.011 and D.02.009, no person shall sell an alimentary paste to which a vitamin or a mineral nutrient set out in column I of any item of the table to this section has been added unless each 100 g of the alimentary paste contains the added vitamin or mineral nutrient in an amount not less than the minimum amount set out in column II of that item and not more than the maximum amount set out in column III of that item.
(2) No person shall represent an alimentary paste as “enriched” unless the alimentary paste contains added thiamine, riboflavin, niacin, folic acid and iron, in accordance with the table to this section.
TABLE
Item Column I Column II Column III Added Vitamin or Mineral Nutrient Minimum Amount per 100 g of Alimentary Paste Maximum Amount per 100 g of Alimentary Paste 1 Thiamine 0.63 mg 1.50 mg 2 Riboflavin 0.11 mg 0.60 mg 3 Niacin 5.90 mg 7.50 mg 4 Folic Acid 0.20 mg 0.27 mg 5 Pantothenic Acid 1.00 mg 2.00 mg 6 Vitamin B6 0.40 mg 0.80 mg 7 Iron 2.90 mg 4.30 mg 8 Magnesium 150.00 mg 300.00 mg
- SOR/94-37, s. 1
- SOR/94-689, s. 2
- SOR/96-527, s. 2
- SOR/98-550, ss. 4, 5
Breakfast Cereal
B.13.060 Notwithstanding sections D.01.009, D.01.011 and D.02.009, no person shall sell a breakfast cereal to which a vitamin or mineral nutrient set out in Column I of an item of the table to this section has been added, either singly or in any combination, unless each 100 g of the breakfast cereal contains the added vitamin or mineral nutrient in the amount set out in Column II of that item.
TABLE
| Column I | Column II | |
|---|---|---|
| Item | Vitamin or Mineral Nutrient | Amount per 100 g of Breakfast Cereal |
| 1 | Thiamine | 2.0 mg |
| 2 | Niacin | 4.8 mg |
| 3 | Vitamin B6 | 0.6 mg |
| 4 | Folic Acid | 0.06 mg |
| 5 | Pantothenic Acid | 1.6 mg |
| 6 | Magnesium | 160.0 mg |
| 7 | Iron | 13.3 mg |
| 8 | Zinc | 3.5 mg |
- SOR/83-858, s. 1
- SOR/89-145, s. 2
- SOR/98-458, s. 7(F)
DIVISION 14Meat, Its Preparations and Products
B.14.001 In this Division,
- animal
animal means any animal used as food, but does not include marine and fresh water animals; (animal)
- filler
filler means any vegetable material (except tomato or beetroot), milk, egg, yeast or any derivative or combination thereof that is acceptable as food. (agent de remplissage)
- SOR/82-768, s. 39
- SOR/86-875, s. 1
B.14.002 [S]. Meat shall be the edible part of the skeletal muscle of an animal that was healthy at the time of slaughter, or muscle that is found in the tongue, diaphragm, heart or oesophagus, and may contain accompanying and overlying fat together with the portions of bone, skin, sinew, nerve and blood vessels that normally accompany the muscle tissue and are not separated from it in the process of dressing, but does not include muscle found in the lips, snout, scalp or ears.
B.14.003 [S]. Meat by-product shall be any edible part of an animal, other than meat, that has been derived from one or more animals that were healthy at the time of slaughter.
B.14.004 Meat, meat by-products or preparations thereof are adulterated if any of the following substances or class of substances are present therein or have been added thereto:
(a) mucous membranes, any organ or portions of the genital system, black gut, spleens, udders, lungs or any other organ or portion of animal that is not commonly sold as an article of food;
(b) preservatives other than those provided for in this Division; or
(c) colour other than annatto, allura red and sunset yellow FCF, where provided for in this Division, and caramel.
- SOR/92-725, s. 2
- SOR/97-516, s. 2
- SOR/2016-74, s. 6(F)
B.14.005 [S]. Prepared meat or a prepared meat by-product shall be any meat or any meat by-product, respectively, whether comminuted or not, to which has been added any ingredient permitted by these Regulations, or which has been preserved, placed in a hermetically-sealed container or cooked, and may contain
(a) a Class II preservative;
(a.1) in the case of prepared hams, shoulders, butts, picnics and backs, gelatin;
(b) in the case of partially defatted pork fatty tissue and partially defatted beef fatty tissue, a Class IV preservative;
(c) where a minimum total protein content or a minimum meat protein content is prescribed in this Division, phosphate salts that do not when calculated as sodium phosphate, dibasic, exceed the maximum level provided therefor in Table XII to section B.16.100 and that are one or more of the following phosphate salts, namely,
(i) sodium acid pyrophosphate,
(ii) sodium hexametaphosphate,
(iii) sodium phosphate, dibasic,
(iv) sodium phosphate, monobasic,
(v) sodium pyrophosphate, tetrabasic,
(vi) sodium tripolyphosphate,
(vii) potassium phosphate, monobasic,
(viii) potassium phosphate, dibasic, and
(ix) potassium pyrophosphate, tetrabasic; and
(d) in the case of vacuum-packed sliced roast beef and vacuum-packed sliced cooked ham, Carnobacterium maltaromaticum CB1.
- SOR/94-262, s. 2
- SOR/2010-264, s. 1
- SOR/2011-280, s. 1
B.14.006 Powdered fully hydrogenated cottonseed oil may be applied as a release agent to the surface of meat, meat by-product, prepared meat, prepared meat by-product, extended meat product and simulated meat product in an amount not greater than 0.25% of the product.
- SOR/2010-142, s. 59(F)
- SOR/2022-168, s. 42
B.14.007 [S]. Meat Binder or (naming the meat product) Binder shall be a filler with any combination of salt, sweetening agents, spices or other seasonings (except tomato), egg, egg albumen, and
(a) where sold for use in preserved meat or preserved meat by-product, may contain any of ascorbic acid, calcium ascorbate, erythorbic acid, iso-ascorbic acid, potassium nitrate, potassium nitrite, sodium ascorbate, sodium carbonate, sodium erythorbate, sodium iso-ascorbate, sodium nitrate and sodium nitrite, provided that these nitrates and nitrites, if any, are packaged separately from any spice or seasoning.
(b) where sold for use in prepared meat or meat by-product in which a gelling agent is a permitted ingredient, may contain a gelling agent;
(c) where sold for use in fresh, uncooked sausage, may contain artificial maple flavour; and
(d) may contain an anticaking agent.
- SOR/80-13, s. 1
- SOR/82-913, s. 1
- SOR/86-875, s. 2(F)
- SOR/2010-143, s. 9
- SOR/2017-18, s. 3(F)
B.14.008 No person shall sell a meat binder, filler or preparations for pumping pickle, cover pickle or dry cure represented for use in meat products unless the label thereof carries directions for use that when followed will produce a food that will comply with the requirements of section B.14.030 insofar as the filler is concerned and the food will not contain food additives in excess of the maximum levels of use prescribed by these Regulations.
- SOR/84-300, s. 44(E)
B.14.009 [S]. Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product may contain
(a) Class I preservatives if the nitrate or nitrite salts or both are packaged separately from any spice or seasoning;
(b) citric acid, sodium citrate or vinegar;
(c) sweetening agents, including maple sugar and maple syrup;
(d) liquid smoke flavour, liquid smoke flavour concentrate, salt, seasonings, spices, spice extracts, spice oils or spice oleoresins;
(e) sodium bicarbonate, sodium hydroxide or potassium hydroxide;
(f) in the case of pumping pickle for cured pork, beef and lamb cuts, disodium phosphate, monosodium phosphate, sodium hexametaphosphate, sodium tripolyphosphate, tetrasodium pyrophosphate and sodium acid pyrophosphate in such amount calculated as disodium phosphate, as will result in the finished product containing not more than 0.5 per cent added phosphate;
(g) in the case of pumping pickle for cured beef cuts, enzymes, if the principal display panel of the label of the cured beef carries, immediately preceding or following the common name, the statement “Tenderized with (naming the proteolytic enzyme or enzymes)”;
(h) in the case of dry cure, an anticaking agent or a humectant; and
(i) in the case of pumping pickle
(i) for cured pork hams, shoulders and backs, artificial maple flavour, and
(ii) for cured pork bellies, artificial maple flavour and an orange flavour that meets the standard prescribed in section B.10.005.
- SOR/79-251, s. 4
- SOR/80-13, s. 2
- SOR/82-596, s. 1
- SOR/88-336, s. 3
- SOR/94-567, s. 1
- SOR/2010-143, s. 10(F)
- SOR/2017-18, s. 4(F)
- SOR/2018-69, s. 4(F)
B.14.010 No person shall sell as food a dead animal or any part thereof.
B.14.011 No person shall sell as food, meat, meat by-products, preparations containing meat or meat derivatives obtained, prepared or manufactured from a dead animal.
B.14.012 For the purpose of Sections B.14.010 and B.14.011, dead animal means a dead animal that
(a) was not killed for the purpose of food in accordance with commonly accepted practice of killing animals for the purpose of food, which shall include exsanguination; or
(b) was affected with disease at the time it was killed.
B.14.013 and B.14.014 [Repealed, SOR/97-148, s. 3]
Meat, Meat By-Products
B.14.015 [S]. Regular Ground Beef shall be beef meat processed by grinding and shall contain not more than 30 per cent beef fat, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981.
- SOR/82-768, s. 40
B.14.015A [S]. Medium Ground Beef shall be beef meat processed by grinding and shall contain not more than 23 per cent beef fat, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981.
- SOR/82-768, s. 40
B.14.015B [S]. Lean Ground Beef shall be beef meat processed by grinding and shall contain not more than 17 per cent beef fat, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981.
- SOR/82-768, s. 40
B.14.015C No person shall sell ground beef that contains more than 30 per cent beef fat, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981.
- SOR/82-768, s. 40
B.14.016 No person shall sell horse-meat or horse-meat by-product, or any food containing horse-meat or horse-meat by-product unless
(a) it is labelled as such when offered or exposed for sale; and
(b) when in package form, the principal display panel of the label carries a declaration of the presence of horse-meat or of horse-meat by-product in type at least as legible and conspicuous as any other type upon such principal display panel.
- SOR/88-336, s. 3
B.14.017 [Repealed, SOR/2003-292, s. 2]
B.14.018 (1) Subject to subsection (2), if a carcass of beef or veal, or a portion of a carcass of beef or veal that weighs 7 kg or more, is advertised for sale, the advertisement shall indicate
(a) in the case of a carcass other than an imported carcass, the grade that was assigned to the carcass under the Safe Food for Canadians Act or a provincial law;
(b) in the case of an imported beef carcass, the grade that was assigned to the carcass under the Safe Food for Canadians Act or a provincial law or the grade that was assigned to the carcass by a grading authority established under the laws of the country from which the carcass was imported;
(c) in the case of an imported veal carcass, the grade that was assigned to the carcass by a grading authority established under the laws of the country from which the carcass was imported; and
(d) in the case of a beef carcass, the yield class, if any, that was assigned to the carcass under the Safe Food for Canadians Act.
(2) Where, in the case of a carcass referred to in subsection (1), no grade has been assigned thereto as described in that subsection and the carcass or portion thereof that weighs 7 kg or more is advertised for sale, the advertisement shall clearly indicate that the carcass has not been graded.
- SOR/92-626, s. 15
- SOR/2003-6, s. 79
- SOR/2018-108, s. 398
B.14.019 (1) Where a carcass of beef, veal, pork or lamb or a portion thereof that weighs 7 kg or more is advertised for sale and a selling price is stated in the advertisement, the advertisement shall
(a) contain the words “price per kilogram is based on carcass weight before cutting, boning and trimming” or the words “price per kilogram is based on the weight of the meat after cutting, boning and trimming”, whichever words are applicable; and
(b) where in addition to the selling price a charge is payable for cutting, boning, trimming, wrapping or freezing the carcass or portion thereof, indicate
(i) the amount of the additional charge, and
(ii) where the additional charge is payable on a price per unit weight basis, whether the additional charge is based on the weight of the carcass or portion thereof before or after the carcass has been cut, boned and trimmed.
(2) Any information required by subsection (1) to appear in an advertisement shall be located therein immediately adjacent to the selling price stated therein, without any intervening written, printed or graphic matter.
- SOR/92-626, s. 15
- SOR/95-548, s. 5(F)
B.14.020 [S]. Solid cut meat shall be
(a) a whole cut of meat; or
(b) a product consisting of pieces of meat of which at least 80 per cent weigh at least 25 g each.
- SOR/94-262, s. 3
B.14.021 (1) No person shall sell solid cut meat to which phosphate salts or water has been added unless
(a) in the case of meat, other than side bacon, Wiltshire bacon, pork jowls, salt pork and salt beef, the meat
(i) where cooked, contains a meat protein content of not less than 12 per cent, and
(ii) where uncooked, contains a meat protein content of not less than 10 per cent; and
(b) that meat contains, phosphate salts that do not when calculated as sodium phosphate, dibasic, exceed the maximum level provided therefor in Table XII to section B.16.100 and that are one or more of the following phosphate salts, namely,
(i) sodium acid pyrophosphate,
(ii) sodium hexametaphosphate,
(iii) sodium phosphate, dibasic,
(iv) sodium phosphate, monobasic,
(v) sodium pyrophosphate, tetrabasic,
(vi) sodium tripolyphosphate,
(vii) potassium phosphate, monobasic,
(viii) potassium phosphate, dibasic, and
(ix) potassium pyrophosphate, tetrabasic.
(2) A bone or a visible fat layer shall not be included in any calculation used to determine meat protein content for the purposes of paragraph (1)(a).
- SOR/94-262, s. 3
B.14.022 No person shall sell mechanically tenderized beef unless
(a) the beef is identified as mechanically tenderized when it is offered or exposed for sale; and
(b) the beef has a label on it with a principal display panel that, subject to section B.01.012, includes all of the following information:
(i) in type at least as legible and prominent as that of the common name,
(A) the expression “mechanically tenderized” in the English version of the label, and
(B) the expression “attendri mécaniquement”, with any necessary grammatical modifications, in the French version of the label, and
(ii) in type at least as legible and prominent as that of any other information other than the common name,
(A) in the English version of the label,
(I) the message “Cook to a minimum internal temperature of 63°C (145°F).”, and
(II) in the case of steak, the additional message “Turn steak over at least twice during cooking.”, and
(B) in the French version of the label,
(I) the message “Faire cuire jusqu’à ce que la température interne atteigne au moins 63 °C (145 °F).”, and
(II) in the case of steak, the additional message “Retourner le bifteck au moins deux fois durant la cuisson.”.
- SOR/2014-99, s. 2
Prepared Meats, Prepared Meat By-Products
B.14.030 (1) Subject to subsections (2) and (3) and section B.14.030A, no person shall sell a prepared meat or prepared meat by-product with a meat protein content of less than 1.5 percentage points below the total protein requirement for that food.
(2) Subsection (1) does not apply to an extended meat product.
(3) Where gelatin is an ingredient of a prepared meat or prepared meat by-product, that gelatin shall not be included when calculating the total protein content of the prepared meat or prepared meat by-product.
- SOR/78-637, s. 3
- SOR/79-251, s. 5(F)
- SOR/80-13, s. 3
- SOR/82-768, s. 41
- SOR/86-875, s. 3
B.14.030A For the purposes of sections B.14.030, B.14.032, B.14.033, B.14.035, B.14.074, B.14.075, B.14.076 and B.14.077, where any of the non-meat ingredients listed in paragraphs B.14.032A(a) to (g) are present in a prepared meat or prepared meat by-product in separate identifiable pieces or chunks in any amount sufficient to differentiate those ingredients from the prepared meat or prepared meat by-product, those ingredients shall not be included when calculating the fat or protein content of the prepared meat or prepared meat by-product.
- SOR/86-875, s. 3
B.14.031 [S]. Preserved Meat or Preserved Meat By-product shall be cooked or uncooked meat or meat by-product that is salted, dried, pickled, corned, cured or smoked, may be glazed and may contain
(a) a Class I preservative;
(a.1) a Class II preservative;
(b) sweetening agents;
(c) spices and seasonings, except tomato;
(d) vinegar;
(e) alcohol;
(f) smoke flavouring or artificial smoke flavouring;
(g) in the case of cured pork hams, shoulders, backs and bellies, artificial maple flavour;
(gg) in the case of cured pork bellies, an added orange flavour that meets the standard prescribed in section B.10.005;
(h) in the case of cured pork, beef and lamb cuts prepared with the aid of pumping pickle, disodium phosphate, monosodium phosphate, sodium hexametaphosphate, sodium tripolyphosphate, tetrasodium pyrophosphate and sodium acid pyrophosphate in such amount calculated as disodium phosphate, as will result in the finished product containing not more than 0.5 per cent added phosphate;
(i) in the case of tocino, annatto in such amount as will result in the finished product containing not more than 0.1% annatto; and
(j) in the case of vacuum-packed sliced cooked ham, Carnobacterium maltaromaticum CB1.
- SOR/79-251, s. 6
- SOR/80-13, s. 4
- SOR/82-596, s. 2
- SOR/84-300, s. 45(E)
- SOR/88-336, ss. 2, 3
- SOR/92-725, s. 3
- SOR/97-151, s. 24
- SOR/2010-264, s. 2
- SOR/2011-280, s. 2
- SOR/2016-305, s. 57
- SOR/2017-18, s. 5(F)
- SOR/2018-69, s. 5(F)
B.14.032 [S]. Sausage or Sausage Meat
(a) shall be fresh or preserved comminuted meat;
(b) may be enclosed in a casing or have an edible coating;
(c) may be dipped in vinegar, smoked, cooked or dried;
(d) may contain
(i) animal fat,
(ii) filler,
(iii) beef tripe,
(iv) liver,
(v) fresh or frozen beef and pork blood,
(vi) sweetening agents,
(vii) salt and spices,
(viii) seasoning, other than tomato,
(ix) lactic acid producing starter culture,
(x) meat binder,
(xi) beef and pork blood plasma,
(xi.1) a Class II preservative,
(xii) in the case of preserved comminuted meat, smoke flavouring or artificial smoke flavouring,
(xiii) if cooked
(A) glucono delta lactone,
(B) partially defatted beef fatty tissue or partially defatted pork fatty tissue, and
(C) a dried skim milk product, obtained from skim milk by the reduction of its calcium content and a corresponding increase in its sodium content, in an amount not exceeding three per cent of the finished food,
(xiv) in the case of fresh uncooked sausage, artificial maple flavour or apple powder as a flavouring ingredient,
(xv) in the case of dry sausage or dry sausage meat, glucono delta lactone,
(xvi) in the case of longaniza,
(A) annatto in such amount as will result in the finished product containing not more than 0.1% annatto,
(B) allura red in such amount as will result in the finished product containing not more than 80 parts per million allura red, and
(C) sunset yellow FCF in such amount as will result in the finished product containing not more than 20 parts per million sunset yellow FCF,
(xvii) in the case of vacuum-packed wieners, Carnobacterium maltaromaticum CB1, and
(xviii) in the case of sausage having an edible coating, calcium chloride or calcium lactate;
(e) shall contain, in the case of a product sold as fresh sausage, not more than 40 per cent fat, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981;
(f) shall have, if cooked, a total protein content of not less than 11 per cent;
(g) shall have, in the case of fresh uncooked sausage and fresh uncooked sausage meat, a total protein content of not less than nine per cent.
- SOR/80-13, s. 5
- SOR/82-768, s. 42
- SOR/88-336, s. 3
- SOR/92-725, s. 4
- SOR/97-151, s. 25
- SOR/97-516, s. 3
- SOR/2010-264, s. 3
- SOR/2011-280, s. 3
- SOR/2012-111, s. 1
- SOR/2016-305, s. 58
B.14.032A [S]. (naming the prepared meat or prepared meat by-product) with (naming the non-meat ingredients) shall be prepared meat to which has been added other non-meat ingredients including
(a) fruit;
(b) vegetables;
(c) nuts;
(d) cheese or processed cheese;
(e) macaroni;
(f) pickles; or
(g) olives.
- SOR/84-300, s. 46
B.14.032AA Where the non-meat ingredients referred to in section B.14.032A are added to prepared meat in such a manner that they are not present in the final product in separate identifiable pieces or chunks, the final product shall meet the total protein content requirement established for (naming the prepared meat or prepared meat by-product) referred to in section B.14.032A.
- SOR/86-875, s. 4
B.14.033 [S]. Potted Meat, Meat Paste or Meat Spread shall be fresh or preserved meat that is comminuted and cooked, and may contain meat binder, salt, sweetening agents, spices, other seasonings, a gelling agent, sodium acetate and sodium diacetate and shall have a total protein content of not less than 9%.
- SOR/80-13, s. 6
- SOR/2011-280, s. 4
- SOR/2017-18, s. 19(F)
B.14.034 [S]. Potted Meat By-product, Meat By-product Paste or Meat By-product Spread shall be a food that
(a) consists, wholly or in part, of meat by-products and conforms to the standard prescribed for potted meat; and
(b) in the case of liverpaste or liverwurst spread, may contain wheat germ and yeast.
- SOR/78-637, s. 4
- SOR/80-13, s. 7
- SOR/86-875, s. 5
B.14.035 [S]. Meat Loaf, Meat Roll, Meat Lunch or Luncheon Meat shall be fresh or preserved meat that is comminuted, cooked and pressed into shape, and may contain a dried skim milk product obtained from skim milk by the reduction of its calcium content and a corresponding increase in its sodium content, in an amount not exceeding 3% of the finished food, as well as filler, meat binder, salt, sweetening agents, glucono delta lactone, spices, other seasonings, milk, eggs, a gelling agent, sodium acetate, sodium diacetate and partially defatted beef fatty tissue or partially defatted pork fatty tissue, and shall have a total protein content of not less than 11%.
- SOR/80-13, s. 8
- SOR/2011-280, s. 5
- SOR/2017-18, s. 19(F)
B.14.036 [S]. Meat By-product Loaf or Meat and Meat By-product Loaf shall be the food consisting, wholly or in part, of meat by-products and shall otherwise conform to the standard prescribed for meat loaf.
B.14.037 (1) [S]. Headcheese
(a) shall be comminuted cooked meat or comminuted cooked preserved meat,
(b) shall not contain
(i) less than 50 per cent head meat, or
(ii) skin, other than that naturally adherent to any pork meat used,
(c) may contain scalps, snouts, beef tripe, salt, spices, seasoning or an added gelling agent, and
(d) may contain
(i) a Class I preservative, and
(ii) a Class II preservative.
(2) For the purpose of subsection (1), scalps and snouts are considered to be head meat.
- SOR/80-500, s. 5
- SOR/2011-280, s. 6
- SOR/2017-18, s. 19(F)
B.14.038 [S]. Brawn shall be headcheese, except that it need not contain 50 per cent head meat.
B.14.039 [Repealed, SOR/2022-143, s. 26]
B.14.040 Subject to section B.14.032 and sections B.14.033 to B.14.036, no person shall sell a food that consists of a mixture of ground meat and filler, ground meat by-product and filler or ground meat, ground meat by-product and filler, unless that food
(a) has a total protein content of not less than 13 per cent;
(b) has a fat content of not more than 40 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case of a mixture containing pork meat or pork meat by-product or both; and
(c) has a fat content of not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case of any other meat mixture.
- SOR/79-251, s. 7(F)
- SOR/82-768, s. 43
B.14.041 Subject to section B.14.032 and sections B.14.033 to B.14.036, no person shall sell a food that consists of a mixture of ground meat and spices and seasonings, ground meat by-product and spices and seasonings, ground meat, ground meat by-product and spices and seasonings or ground meat and ground meat by-product, unless that food
(a) has a total protein content of not less than 16 per cent;
(b) has a fat content of not more than 40 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case of a mixture containing pork meat or pork meat by-product or both; and
(c) has a fat content of not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case of any other meat mixture.
- SOR/79-251, s. 7(F)
- SOR/82-768, s. 44
Meat Derivatives
B.14.061 [S]. Edible Bone Meal or Edible Bone Flour shall be the food prepared by grinding dry, defatted bones, obtained from animals healthy at the time of slaughter and shall contain
(a) not less than 85 per cent ash, as determined by official method FO-34, Determination of Ash in Edible Bone Meal or Edible Bone Flour, October 15, 1981.
(b) and (c) [Repealed, SOR/97-148, s. 4]
- SOR/82-768, s. 45
- SOR/97-148, s. 4
B.14.062 [S]. (1) Gelatin or Edible Gelatin
(a) shall be the purified food obtained by the processing of skin, ligaments or bones of animals;
(b) shall contain not less than 82 per cent ash-free solids, when tested by official method FO-35, Determination of Ash-Free Solids in Gelatin, October 15, 1981;
(c) shall be free from objectionable taste and offensive odour when 2.5 grams thereof are dissolved in 100 millilitres of warm water;
(d) [Repealed, SOR/97-148, s. 5]
(e) shall not contain any residues of hydrogen peroxide where it has been used in the course of manufacture; and
(f) may contain
(i) not more than 2.6 per cent ash on a dry basis,
(ii) not more than 500 parts per million of sulphurous acid, including the salts thereof, calculated as sulphur dioxide, and
(iii) where intended for use in the manufacture of marshmallow, sodium hexametaphosphate or sodium lauryl sulphate.
(2) No person shall use, in the course of manufacturing gelatin or edible gelatin,
(a) acidic or basic compounds other than acetic acid, ammonium hydroxide, citric acid, fumaric acid, hydrochloric acid, lime, magnesium hydroxide, phosphoric acid, sodium carbonate, sodium hydroxide, sodium sulphide, sulphuric acid, sulphurous acid or tartaric acid; or
(b) filtering and clarifying agents other than activated carbon, alumina, aluminum sulphate, calcium phosphate, dibasic, cellulose, diatomaceous earth, perlite, strongly acidic cation exchange resin in the hydrogen ion form or basic anion exchange resins in the chloride ion or free base ion forms.
- SOR/78-401, s. 2(E)
- SOR/78-874, s. 1
- SOR/80-501, s. 3
- SOR/82-768, s. 46
- SOR/97-148, s. 5
Meat Stews
B.14.063 For the purposes of sections B.14.064 to B.14.068, stew meat means meat that contains when raw not more than
(a) 25 per cent fat, in the case of meat in meat ball stews; and
(b) 20 per cent fat, in the case of meat in other stews.
- SOR/78-874, s. 2
B.14.064 [S]. Vegetable Stew with (naming the meat)
(a) shall contain vegetables and the named meat in the following amounts, calculated as raw ingredients:
(i) 12 per cent or more stew meat, and
(ii) 38 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 2
B.14.065 [S]. (naming the meat) Stew
(a) shall contain vegetables and the named meat in the following amounts, calculated as raw ingredients:
(i) 20 per cent or more stew meat, and
(ii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 2
B.14.066 [S]. Irish Stew
(a) shall contain mutton, lamb or beef, singly or in any combination, and vegetables, in the following amounts, calculated as raw ingredients:
(i) 20 per cent or more stew meat,
(ii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 2
B.14.067 [S]. Meat Ball Stew
(a) shall contain vegetables and meat balls in the following amounts, calculated as raw ingredients:
(i) 22 per cent or more meat balls,
(ii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 2
B.14.068 [S]. Specialty Meat Stews
(a) shall contain meat and vegetables in the following amounts, calculated as raw ingredients:
(i) 25 per cent or more stew meat,
(ii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 2
Meat Specialties
B.14.070 [S]. Wieners and Beans or Wieners with Beans shall be the food prepared from dried beans and wieners, may contain sauce, seasoning, spices and a sweetening agent and shall contain not less than 25 per cent wieners, as determined by official method FO-36, Determination of Wiener Content of Meat Specialties, October 15, 1981.
- SOR/82-768, s. 47
B.14.071 [S]. Beans and Wieners or Beans with Wieners shall be the food prepared from dried beans and wieners, may contain sauce, seasoning, spices and a sweetening agent and shall contain not less than 10 per cent wieners, as determined by official method FO-36, Determination of Wiener Content of Meat Specialties, October 15, 1981.
- SOR/82-768, s. 47
Sale of Barbecued, Roasted or Broiled Meat or Meat By-Products
B.14.072 No person shall sell meat or a meat by-product that has been barbecued, roasted or broiled and is ready for consumption unless the cooked meat or meat by-product
(a) at all times
(i) has a temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher, or
(ii) has been stored at an ambient temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher; and
(b) carries on the principal display panel of the label a statement to the effect that the food must be stored at a temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher.
- SOR/78-403, s. 4(F)
- SOR/88-336, s. 3
Meat Product Extender
B.14.073 No person shall sell a meat product extender intended to be used in a food consisting of a mixture described in section B.14.074, B.14.075, B.14.076, B.14.077 or B.14.078, unless that extender
(a) has, in the rehydrated state,
(i) a total protein content of not less than 16 per cent, and
(ii) a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(b) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(c) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 48
Extended Meat Products
B.14.074 Subject to sections B.14.075 to B.14.079, no person shall sell a food that consists of a mixture of meat product and meat product extender, unless that food
(a) has a total protein content of not less than 16 per cent;
(b) has a fat content of not more than 25 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 49
B.14.075 No person shall sell a food that consists of a mixture of meat product and meat product extender and that resembles fresh sausage, unless that food
(a) has a total protein content of not less than nine per cent;
(b) has a fat content of not more than 40 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 50
- SOR/84-300, s. 47(F)
B.14.076 No person shall sell a food that consists of a mixture of meat product and meat product extender and that resembles cooked sausage, meat loaf, meat by-product loaf, meat roll, meat lunch, or luncheon meat, unless that food
(a) has a total protein content of not less than 11 per cent;
(b) has a fat content of not more than 25 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 51
B.14.077 No person shall sell a food that consists of a mixture of meat product and meat product extender and that resembles potted meat, potted meat by-product, meat paste, meat by-product paste, meat spread, or meat by-product spread, unless that food
(a) has a total protein content of not less than nine per cent;
(b) has a fat content of not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 52
- SOR/84-300, s. 48(E)
B.14.078 No person shall sell a food that consists of a mixture of meat product and meat product extender and that resembles regular ground beef, medium ground beef or lean ground beef, unless that food
(a) has a total protein content of not less than 16 per cent;
(b) has a fat content of
(i) not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being regular,
(ii) not more than 23 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being medium, or
(iii) not more than 17 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being lean; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 53
B.14.079 No person shall sell a food that consists of a mixture of meat product, filler and meat product extender, unless that food
(a) has a total protein content of not less than 13 per cent;
(b) has a fat content of not more than 25 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981; and
(c) in respect of the meat product extender, meets the requirements of paragraphs B.14.073(a) to (c).
- SOR/82-768, s. 54
Simulated Meat Products
B.14.085 Subject to sections B.14.086 to B.14.090 no person shall sell a simulated meat product unless that product
(a) has, in the rehydrated state,
(i) a total protein content of not less than 16 per cent,
(ii) a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981, and
(iii) a fat content of not more than 25 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981;
(b) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(c) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 55
B.14.086 No person shall sell a simulated meat product that resembles fresh sausage, unless that product
(a) has a total protein content of not less than nine per cent;
(b) has a protein rating of not less than 23, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) has a fat content of not more than 40 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981;
(d) has, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(e) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 56
B.14.087 No person shall sell a simulated meat product that resembles cooked sausage, meat loaf, meat by-product loaf, meat roll, meat lunch, or luncheon meat, unless that product
(a) has a total protein content of not less than 11 per cent;
(b) has a protein rating of not less than 28, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) has a fat content of not more than 25 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981;
(d) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(e) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 57
B.14.088 No person shall sell a simulated meat product that resembles potted meat, potted meat by-product, meat paste, meat by-product paste, meat spread, or meat by-product spread, unless that product
(a) has a total protein content of not less than nine per cent;
(b) has a protein rating of not less than 23, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) has a fat content of not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981;
(d) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each vitamin and mineral nutrient respectively; and
(e) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 58
B.14.089 No person shall sell a simulated meat product that resembles regular ground beef, medium ground beef or lean ground beef, unless that product
(a) has a total protein content of not less than 16 per cent;
(b) has a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) has a fat content of
(i) not more than 30 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being regular,
(ii) not more than 23 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being medium, or
(iii) not more than 17 per cent, as determined by official method FO-33, Determination of Fat in Meat and Simulated Meat Products, October 15, 1981, in the case where the product is represented as being lean;
(d) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient, respectively; and
(e) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 59
B.14.090 No person shall sell a simulated meat product that resembles side bacon, unless that product
(a) has a total protein content of not less than 25 per cent;
(b) has a protein rating of not less than 20, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to this Division in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(d) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 60
TABLE
| Item No. | Column I | Column II |
|---|---|---|
| Vitamin or Mineral Nutrient | Amount per gram of protein | |
C.1  | Copper | 4.4 micrograms |
| F.1 | Folic Acid | 0.45 microgram |
| I.1 | Iron | 0.25 milligram |
| M.1 | Magnesium | 1.1 milligrams |
| N.1 | Niacin | 0.34 milligram |
| P.1 | Pantothenic Acid | 0.04 milligram |
| P.2 | Potassium | 20 milligrams |
| P.3 | Pyridoxine | 0.02 milligram |
| R.1 | Riboflavin | 0.01 milligram |
| T.1 | Thiamine | 0.02 milligram |
| V.1 | Vitamin B12 | 0.08 microgram |
| Z.1 | Zinc | 0.20 milligram |
- SOR/98-458, s. 7(F)
DIVISION 15Adulteration of Food
B.15.001 (1) A food referred to in column 2 of Part 1 of the List of Contaminants and Other Adulterating Substances in Foods is adulterated if the corresponding substance referred to, by name or class, in column 1 is present in or on the food.
(2) A food referred to in column 2 of Part 2 of the List of Contaminants and Other Adulterating Substances in Foods is adulterated if the corresponding substance referred to, by name or class, in column 1 is present in or on the food in an amount that exceeds the maximum level set out in column 3.
(3) If a substance referred to, by name or class, in column 1 in Part 2 the List of Contaminants and Other Adulterating Substances in Foods is present in or on the corresponding food referred to in column 2, the food is, in respect of the presence of the substance, exempt from the application of paragraph 4(1)(a) of the Act if the amount of the substance does not exceed the maximum level set out in column 3.
(4) Subsections (1) to (3) do not apply to a substance that is present in or on a food as
(a) a food additive;
(a.1) a supplemental ingredient;
(b) a pest control product as defined in subsection 2(1) of the Pest Control Products Act or its components or derivatives; or
(c) a veterinary drug or its metabolites.
- SOR/78-404, s. 1
- SOR/79-249, s. 1
- SOR/2016-74, s. 7
- SOR/2022-169, s. 19
B.15.002 (1) Subject to subsection (2), a food is adulterated if
(a) a pest control product as defined in subsection 2(1) of the Pest Control Products Act or its components or derivatives, for which no maximum residue limit has been specified under sections 9 or 10 of that Act for that food, are present in or on the food, singly or in any combination, in an amount exceeding 0.1 part per million; or
(b) an agricultural chemical or its components or derivatives, other than a pest control product as defined in subsection 2(1) of the Pest Control Products Act or its components or derivatives, are present in or on the food, singly or in any combination, in an amount exceeding 0.1 part per million.
(2) A food is exempt from paragraph 4(1)(d) of the Act if the following agricultural chemicals, or their components or derivatives, are the only agricultural chemicals, or components or derivatives of agricultural chemicals, that are present in or on the food, singly or in any combination:
(a) a fertilizer;
(b) an adjuvant or a carrier of an agricultural chemical;
(c) an inorganic bromide salt;
(d) silicon dioxide;
(e) sulphur;
(f) viable spores of Bacillus thuringiensis Berliner; or
(g) Kaolin.
(3) Subsection (2) does not apply to a food if there is present in or on the food an agricultural chemical, or a component or derivative of that agricultural chemical, referred to in that subsection that is a pest control product as defined in subsection 2(1) of the Pest Control Products Act, or a component or derivative of that product, in respect of which a maximum residue limit has been specified under sections 9 or 10 of that Act for that food.
(4) [Repealed, SOR/2008-181, s. 3]
- SOR/78-404, s. 1
- SOR/79-249, s. 1
- SOR/81-83, s. 2
- SOR/97-313, s. 2
- SOR/98-98, s. 1
- SOR/2005-67, s. 1
- SOR/2008-181, s. 3
- SOR/2008-182, s. 2
B.15.003 [Repealed, SOR/2016-74, s. 8]
DIVISION 16Food Additives
B.16.001 A quantitative statement of the amount of each additive present or directions for use that, if followed, will produce a food that will not contain such additives in excess of the maximum levels of use prescribed by these Regulations shall be shown, grouped together with the list of ingredients, of any substance or mixture of substances for use as a food additive.
B.16.002 A request that a food additive be added to or a change made in the Tables following section B.16.100 shall be accompanied by a submission to the Minister in a form, manner and content satisfactory to him and shall include
(a) a description of the food additive, including its chemical name and the name under which it is proposed to be sold, its method of manufacture, its chemical and physical properties, its composition and its specifications and, where that information is not available, a detailed explanation;
(b) a statement of the amount of the food additive proposed for use, and the purpose for which it is proposed, together with all directions, recommendations and suggestions for use;
(c) where necessary, in the opinion of the Minister, an acceptable method of analysis suitable for regulatory purposes that will determine the amount of the food additive and of any substance resulting from the use of the food additive in the finished food;
(d) data establishing that the food additive will have the intended physical or other technical effect;
(e) detailed reports of tests made to establish the safety of the food additive under the conditions of use recommended;
(f) data to indicate the residues that may remain in or upon the finished food when the food additive is used in accordance with good manufacturing practice;
(g) a proposed maximum limit for residues of the food additive in or upon the finished food;
(h) specimens of the labelling proposed for the food additive; and
(i) a sample of the food additive in the form in which it is proposed to be used in foods, a sample of the active ingredient, and, on request a sample of food containing the food additive.
- SOR/2018-69, s. 27
B.16.003 The Minister shall, within 90 days after the filing of a submission in accordance with section B.16.002, notify the person filing the submission whether or not it is his intention to recommend to the Governor-in-Council that the said food additive be so listed and the detail of any listing to be recommended.
B.16.004 [Repealed, SOR/97-148, s. 6]
B.16.006 Paragraph B.01.042(c) and paragraph B.01.043(a) do not apply to spices, seasonings, flavouring preparations, essential oils, oleoresins and natural extractives.
B.16.007 No person shall sell a food containing a food additive other than a food additive provided for in sections B.01.042, B.01.043 and B.25.062.
- SOR/87-640, s. 5
B.16.008 [Repealed, SOR/88-418, s. 5]
B.16.100 No person shall sell any substance as a food additive unless the food additive is listed in one or more of the following Tables:
TABLE I
Food Additives That May Be Used as Anticaking Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| C.1 | Calcium Aluminum Silicate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Unstandardized dry mixes | (3) Good Manufacturing Practice | ||
| C.2 | Calcium Phosphate tribasic | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Dry cure | (3) Good Manufacturing Practice | ||
| (4) Unstandardized dry mixes | (4) Good Manufacturing Practice | ||
| (5) Oil soluble annatto | (5) Good Manufacturing Practice | ||
| (6) Icing sugar | (6) If used either singly or in combination with Calcium Silicate, Magnesium Carbonate, Magnesium Silicate, Magnesium Stearate, Silicon Dioxide or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| C.3 | Calcium Silicate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Baking Powder | (3) 5.0% | ||
| (4) Dry cure | (4) Good Manufacturing Practice | ||
| (5) Unstandardized dry mixes | (5) Good Manufacturing Practice | ||
| (6) Icing sugar | (6) If used either singly or in combination with Calcium Phosphate tribasic, Magnesium Carbonate, Magnesium Silicate, Magnesium Stearate, Silicon Dioxide or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| (7) Meat Binder or (naming the meat product) Binder | (7) 1.0% | ||
| (8) Grated or shredded (naming the variety) cheese; Grated or shredded cheddar cheese; Unstandardized grated or shredded cheese preparations | (8) If used singly or in combination with microcrystalline cellulose or cellulose, the total amount not to exceed 2.0% | ||
| (9) Dried egg-white (dried albumen); Dried whole egg; Dried whole egg mix; Dried yolk; Dried yolk mix | (9) 2.0% | ||
| C.4 | Calcium Stearate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Unstandardized dry mixes | (3) Good Manufacturing Practice | ||
| C.5 | Cellulose | Grated or shredded (naming the variety) cheese; Grated or shredded cheddar cheese; Unstandardized grated or shredded cheese preparations | If used singly or in combination with calcium silicate or microcrystalline cellulose, the total amount not to exceed 2.0% |
| M.1 | Magnesium Carbonate | (1) Salt (except when used in preparations of Meat and Meat By-products of Division 14) | (1) 1.0%, except in the case, of fine grained salt 2.0%, in accordance with the requirements of paragraph B.17.001(1)(a) |
| (2) Garlic salt, Onion salt (except when used in preparations of Meat and Meat By-products of Division 14) | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Unstandardized Dry Mixes (Except when used in preparations of Meat and Meat by-products of Division 14) | (3) Good Manufacturing Practice | ||
| (4) Icing sugar | (4) If used either singly or in combination with Calcium Phosphate tribasic, Calcium Silicate, Magnesium Silicate, Magnesium Stearate, Silicon Dioxide or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| M.2 | Magnesium Oxide | Unstandardized dry mixes (Except when used in preparations of Meat and Meat by-products of Division 14) | Good Manufacturing Practice |
| M.3 | Magnesium Silicate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Unstandardized dry mixes | (3) Good Manufacturing Practice | ||
| (4) Icing sugar | (4) If used either singly or in combination with Calcium Phosphate tribasic, Calcium Silicate, Magnesium Carbonate, Magnesium Silicate, Silicon Dioxide or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| M.4 | Magnesium Stearate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Garlic salt; Onion salt | (2) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (3) Unstandardized dry mixes | (3) Good Manufacturing Practice | ||
| (4) Icing sugar | (4) If used either singly or in combination with Calcium Phosphate tribasic, Calcium Silicate, Magnesium Carbonate, Magnesium Silicate, Silicon Dioxide or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| M.5 | Microcrystalline Cellulose | Grated or shredded (naming the variety) cheese; Grated or shredded cheddar cheese; Unstandardized grated or shredded cheese preparations | If used singly or in combination with calcium silicate or cellulose, the total amount not to exceed 2.0% |
| P.1 | Propylene Glycol | Salt | 0.035% |
| S.1 | Silicon Dioxide | (1) Garlic salt; Onion salt | (1) 1.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively |
| (2) Celery Salt; Celery Pepper | (2) 0.5% | ||
| (3) Unstandardized dry mixes | (3) Good Manufacturing Practice | ||
| (4) Icing sugar | (4) If used either singly or in combination with Calcium Phosphate tribasic, Calcium Silicate, Magnesium Carbonate, Magnesium Silicate, Magnesium Stearate or Sodium Aluminum Silicate the total must not exceed 1.5% | ||
| (5) Foods sold in tablet form | (5) Good Manufacturing Practice | ||
| (6) Cayenne Pepper; Chili pepper; Chili Powder; Paprika; Red Pepper | (6) 2.0% | ||
| (7) Salt | (7) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) | ||
| S.2 | Sodium Aluminum Silicate | (1) Salt | (1) 1.0%, except in the case of fine grained salt 2.0%, in accordance with the requirement of paragraph B.17.001(1)(a) |
| (2) Icing sugar | (2) If used either singly or in combination with Calcium Phosphate tribasic, Calcium Silicate, Magnesium Carbonate, Magnesium Silicate, Magnesium Stearate or Silicon Dioxide the total must not exceed 1.5% | ||
| (3) Dried egg-white (dried albumen); Dried whole egg; Dried whole egg mix; Dried yolk; Dried yolk mix | (3) 2.0% | ||
| (4) Garlic salt; Onion salt | (4) 2.0% in accordance with the requirement of paragraphs B.07.020(b) and B.07.027(b) respectively | ||
| (5) Unstandardized dry mixes | (5) Good Manufacturing Practice | ||
| S.3 | Sodium Ferrocyanide, decahydrate | Salt | 13 p.p.m. calculated as anhydrous sodium ferrocyanide |
- SOR/79-662, ss. 3 to 13
- SOR/82-913, s. 4
- SOR/83-410, s. 2
- SOR/84-17, s. 5
- SOR/84-801, s. 2
- SOR/86-1125, s. 1
- SOR/88-534, s. 4
- SOR/91-88, ss. 3, 4
- SOR/93-477, ss. 3 to 5
- SOR/94-689, s. 2(F)
- SOR/97-191, s. 3
- SOR/2010-94, s. 8(E)
- SOR/2010-143, ss. 11, 12
TABLE II
Food Additives That May Be Used as Bleaching, Maturing and Dough Conditioning Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acetone Peroxide | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| A.1A | [Repealed, SOR/79-660, s. 3] | ||
| A.2 | Ammonium Persulphate | (1) Flour; Whole wheat flour | (1) 250 p.p.m. |
| (2) Bread | (2) 100 p.p.m. of flour | ||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | ||
| A.2A | Ascorbic Acid | (1) Bread; Flour; Whole wheat flour | (1) 200 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 200 p.p.m. of flour | ||
| A.3 | [Repealed, SOR/79-660, s. 4] | ||
| A.3A | [Repealed, SOR/79-660, s. 4] | ||
| A.4 | Azodicarbonamide | Bread; Flour; Whole wheat flour | 45 p.p.m. of flour |
| B.1 | Benzoyl Peroxide | Flour; Whole wheat flour | 150 p.p.m. |
| C.1 | Calcium Iodate | (1) Bread | (1) 45 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 45 p.p.m. of flour | ||
| C.2 | Calcium Peroxide | (1) Bread | (1) 100 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| C.3 | Calcium Stearoyl-2-Lactylate | (1) Bread | (1) 3,750 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 3,750 p.p.m. of flour | ||
| (3) Cake mixes | (3) 0.5% of dry weight of mix | ||
| C.4 | Chlorine | Flour; Whole wheat flour | Good Manufacturing Practice |
| C.5 | Chlorine Dioxide | Flour; Whole wheat flour | Good Manufacturing Practice |
| C.6 | L-Cysteine Hydrochloride | (1) Bread; Flour; Whole wheat flour | (1) 90 p.p.m. |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| P.1 | [Repealed, SOR/94-227, s. 4] | ||
| P.2 | Potassium Iodate | (1) Bread | (1) 45 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 45 p.p.m. of flour | ||
| P.3 | Potassium Persulphate | (1) Bread | (1) 100 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| S.1 | Sodium Stearoyl-2-Lactylate | (1) Bread | (1) 3,750 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 3,750 p.p.m. of flour | ||
| (3) Pancakes and pancake mixes | (3) 0.3% of dry ingredient weight | ||
| (4) Waffles and waffle mixes | (4) 0.3% of dry ingredient weight | ||
| (5) Cake mixes | (5) 0.5% of dry weight of mix | ||
| S.2 | Sodium Stearyl Fumarate | (1) Bread | (1) 5,000 p.p.m. of flour |
| (2) Unstandardized bakery products | (2) 5,000 p.p.m. of flour | ||
| S.3 | Sodium Sulphite | Biscuit dough | 500 p.p.m. calculated as Sulphur Dioxide |
- SOR/79-660, ss. 3, 4
- SOR/87-640, s. 6
- SOR/92-591, s. 2
- SOR/94-227, s. 4
- SOR/94-689, s. 2(F)
- SOR/2005-98, ss. 3, 8(F)
- SOR/2010-41, s. 9(E)
TABLE III
Food Additives That May Be Used as Colouring Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| 1 |
| (1) Apple (or rhubarb) and (naming the fruit) jam; Bread; Butter; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Fish roe (caviar); Ice cream mix; Ice milk mix; Icing sugar; Liqueur; Lobster paste; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; Packaged fish and meat products that are marinated or otherwise cold-processed (Division 21); Pickles; Pineapple marmalade with pectin; Relishes; Sherbet; Smoked fish; Tomato catsup | (1) Good Manufacturing Practice |
| (2) Dried whole egg; Dried yolk; Frozen whole egg; Frozen yolk; Liquid whole egg; Liquid yolk | (2) Good Manufacturing Practice in accordance with paragraphs B.22.034(b) and B.22.035(b) | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| (4) Vegetable fats and oils | (4) Good Manufacturing Practice in accordance with section B.09.001 | ||
| (5) Margarine | (5) Good Manufacturing Practice | ||
| (6) (naming the variety) Cheese; Cheddar cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (6) Good Manufacturing Practice in accordance with the requirements of sections B.08.033, B.08.034, B.08.037, B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3, B.08.041.4, B.08.041.5, B.08.041.6, B.08.041.7 and B.08.041.8. | ||
| (7) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (7) Good Manufacturing Practice | ||
| (8) Longaniza; Tocino | (8) 0.1% in accordance with the requirements of paragraph B.14.031(i) or subparagraph B.14.032(d)(xvi) | ||
| (9) Edible collagen film (iron oxide only) | (9) Good Manufacturing Practice | ||
| (10) Sausage casings (annatto only) | (10) 1.0% (Residues of annatto in sausage prepared with such casings not to exceed 100 p.p.m.) | ||
| (11) Sausage casings (cochineal only) | (11) 0.75% (Residues of cochineal in sausage prepared with such casings not to exceed 75 p.p.m.) | ||
| 1A | ß-apo- 8′-carotenal Ethyl ß-apo- 8′-carotenoate | (1) Apple (or rhubarb) and (naming the fruit) jam; Bread; Butter; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Fish roe (caviar); Ice cream mix; Ice milk mix; Icing sugar; Liqueur; Lobster paste; Margarine; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; Pickles; Pineapple marmalade with pectin; Relishes; Sherbet; Smoked fish; Tomato catsup | (1) 35 p.p.m. |
| (2) Unstandardized foods | (2) 35 p.p.m. | ||
| (3) (naming the variety) Cheese; Cheddar cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (3) 35 p.p.m., in accordance with the requirements of sections B.08.033, B.08.034, B.08.037, B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3, B.08.041.4, B.08.041.5, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (4) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (4) 35 p.p.m. | ||
| 2 | Caramel | (1) Ale; Apple (or rhubarb) and (naming the fruit) jam; Beer; Brandy; Bread; Brown bread; Butter; Cider; Cider vinegar; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Fish roe (caviar); Hollands gin; Honey wine; Ice cream mix; Ice milk mix; Icing sugar; Liqueur; Lobster paste; Malt liquor; Malt vinegar; Mincemeat; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; Pickles; Pineapple marmalade with pectin; Porter; Relishes; Rum; Sherbet; Smoked fish; Stout; Tomato catsup; Whisky; Wine; Wine vinegar | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (3) Good Manufacturing Practice | ||
| (4) Sausage casings | (4) 15% (Residues of caramel in sausage prepared with such casings not to exceed 0.15%) | ||
| (5) Cream cheese spread with (naming the added ingredients) | (5) 1.5% | ||
| 3 |
| (1) Apple (or rhubarb) and (naming the fruit) jam; Bread; Butter; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Fish roe (caviar); Ice cream mix; Ice milk mix; Icing sugar; Liqueur; Lobster paste; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; Pickles; Pineapple marmalade with pectin; Relishes; Sherbet; Smoked fish; Tomato catsup | (1) 300 p.p.m. singly or in combination in accordance with section B.06.002 |
| (2) Unstandardized foods | (2) 300 p.p.m. singly or in combination in accordance with section B.06.002 | ||
| (3) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (3) 300 p.p.m. singly or in combination in accordance with section B.26.002 | ||
| (4) Salted anchovy, salted scad and salted shrimp | (4) 125 p.p.m. in accordance with the requirements of paragraph B.21.021(d) | ||
| (5) Longaniza | (5) 80 p.p.m. allura red in accordance with the requirements of clause B.14.032(d)(xvi)(B) and 20 p.p.m. sunset yellow FCF in accordance with the requirements of clause B.14.032(d)(xvi)(C) | ||
| (6) Sausage casings (sunset yellow FCF only) | (6) 0.15% (Residues of sunset yellow FCF in sausage prepared with such casings not to exceed 15 p.p.m.) | ||
| (7) Cheese-flavoured corn snacks (sunset yellow FCF only) | (7) 600 p.p.m. singly. If used in combination with other colours listed in column I of this item and of item 4 of this table, the maximum level of use is 300 p.p.m. in accordance with paragraph B.06.002(c) | ||
| 4 |
| (1) Apple (or rhubarb) and (naming the fruit) jam; Bread; Butter; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Fish roe (caviar); Ice cream mix; Ice milk mix; Icing sugar; Liqueur; Lobster paste; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; Pickles; Pineapple marmalade with pectin; Relishes; Sherbet; Smoked fish; Tomato catsup | (1) 100 p.p.m. singly or in combination in accordance with section B.06.002 |
| (2) Unstandardized foods | (2) 100 p.p.m. singly or in combination in accordance with section B.06.002 | ||
| (3) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (3) 100 p.p.m. singly or in combination in accordance with section B.06.002 | ||
| (4) Feta cheese (brilliant blue FCF only) | (4) 0.10 p.p.m. | ||
| 5 | Citrus Red No. 2 | Skins of whole oranges | 2 p.p.m. |
| 6 | Ponceau SX | Fruit Peel; Glacé fruits; Maraschino Cherries | 150 p.p.m. |
| 7 | Gold | Alcoholic beverages | Good Manufacturing Practice |
- SOR/79-752, s. 5
- SOR/80-500, s. 6
- SOR/82-596, s. 3
- SOR/84-440, s. 4
- SOR/84-602, s. 1
- SOR/89-198, ss. 6 to 10
- SOR/92-725, s. 5
- SOR/93-466, s. 2
- SOR/94-689, s. 2(F)
- SOR/95-434, s. 1
- SOR/95-493, s. 1
- SOR/97-516, s. 4
- SOR/98-580, s. 1(F)
- SOR/99-96, s. 1
- SOR/2000-50, s. 1
- SOR/2000-146, ss. 1 to 3
- SOR/2007-75, s. 3
- SOR/2010-94, s. 8(E)
- SOR/2010-143, s. 13
- SOR/2011-235, s. 2(F)
- SOR/2011-281, s. 2
- SOR/2012-43, ss. 16 to 20
TABLE IV
Food Additives That May Be Used as Emulsifying, Gelling, Stabilizing and Thickening Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acacia Gum | (1) Cream; French dressing; (naming the flavour) Milk; Mustard pickles; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; Relishes; Salad dressing; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; | (1) Good Manufacturing Practice |
| (2) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Calorie-reduced margarine | (5) 0.5% in accordance with the requirements of section B.09.017 | ||
| (6) Canned asparagus; Canned green beans; Canned wax beans; Canned peas | (6) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| A.1A | Acacia Gum modified with octenyl succinic anhydride (OSA) | (1) French dressing; Icings; Salad dressing; Unstandardized dressings; Unstandardized sauces | (1) 1% |
| (2) Unstandardized beverages | (2) 0.1% | ||
| (3) Unstandardized flavouring preparations | (3) 0.05% | ||
| A.2 | Acetylated Mono-glycerides | Unstandardized foods | Good Manufacturing Practice |
| A.3 | Acetylated Tartaric Acid Esters of Mono- and Di-glycerides | (1) Bread | (1) 6,000 p.p.m. of flour |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Infant formulas based on crystalline amino acids | (3) 0.024% as consumed | ||
| A.4 | Agar | (1) Brawn; Canned (naming the poultry); Cream; Headcheese; Meat binder or (naming the meat product) binder where sold for use in prepared meat or prepared meat by-product in which a gelling agent is a permitted ingredient; Meat by-product loaf; Meat loaf; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jelly with pectin; Potted meat; Potted meat by-product; Prepared fish or prepared meat (Division 21); Relishes | (1) Good Manufacturing Practice |
| (2) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Calorie-reduced margarine | (5) 0.5% in accordance with the requirements of section B.09.017 | ||
| A.5 | Algin | (1) Ale; Beer; Cream; French dressing; Malt liquor; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Porter; Relishes; Salad dressing; Stout | (1) Good Manufacturing Practice |
| (2) Infant formula | (2) 0.03% as consumed. If used in combination with carrageenan or guar gum or both, the total not to exceed 0.03% | ||
| (3) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (3) 0.5% | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Calorie-reduced margarine | (6) 0.5% in accordance with the requirements of section B.09.017 | ||
| (7) Sour cream | (7) 0.5% in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (8) Canned asparagus; Canned green beans; Canned wax beans; Canned peas | (8) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| (9) Infant formula based on isolated amino acids or protein hydrolysates, or both | (9) 0.1% as consumed. If used in combination with carrageenan or guar gum or both, the total not to exceed 0.1% | ||
| (10) Lactose-free infant formula based on milk protein | (10) 0.05% as consumed. If used in combination with carrageenan or guar gum or both, the total not to exceed 0.05% | ||
| A.6 | Alginic Acid | Same foods as listed for Algin | Same levels as prescribed for Algin |
| A.7 | Ammonium Alginate | Same foods as listed for Algin | Same levels as prescribed for Algin |
| A.8 | Ammonium Carrageenan | Same foods as listed for Carrageenan | Same levels as prescribed for Carrageenan |
| A.9 | Ammonium Furcelleran | Same foods as listed for Furcelleran | Same levels as prescribed for Furcelleran |
| A.9A | Ammonium Salt of Phosphorylated Glyceride | (1) Bread; Cream; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Relishes | (1) Good Manufacturing Practice |
| (2) Ice Cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Chocolate products; Cocoa products | (5) 0.7% | ||
| A.10 | Arabino-galactan | Essential oils; Pie filling mixes; Pudding mixes; Unstandardized beverage bases; Unstandardized beverage mixes; Unstandardized dressings | Good Manufacturing Practice |
| B.1 | Baker’s yeast Glycan | Unstandardized foods | Good Manufacturing Practice |
| C.1 | Calcium Alginate | Same foods as listed for Algin | Same levels as prescribed for Algin |
| C.2 | Calcium Carbonate | (1) Unstandardized Foods | (1) Good Manufacturing Practice |
| (2) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (2) Good Manufacturing Practice | ||
| C.3 | Calcium Carrageenan | Same foods as listed for Carrageenan | Same levels as prescribed for Carrageenan |
| C.4 | Calcium Citrate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.5 | Calcium Furcelleran | Same foods as listed for Furcelleran | Same levels as prescribed for Furcelleran |
| C.6 | Calcium Gluconate | Unstandardized foods | Good Manufacturing Practice |
| C.7 | Calcium Glycerophosphate | Unstandardized dessert mixes | Good Manufacturing Practice |
| C.8 | Calcium Hypophosphite | Unstandardized dessert mixes | Good Manufacturing Practice |
| C.9 | Calcium Phosphate, dibasic | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.10 | Calcium Phosphate, tribasic | Unstandardized foods | Good Manufacturing Practice |
| C.11 | Calcium Sulphate | (1) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (1) 0.5% |
| (2) Sherbet | (2) 0.75% | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| (4) Creamed cottage cheese | (4) 0.05% | ||
| (5) Cream for whipping, heat-treated above 100°C | (5) 0.005% | ||
| (6) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (6) 0.06% | ||
| C.12 | Calcium Tartrate | Unstandardized foods | Good Manufacturing Practice |
| C.13 | Carboxymethyl Cellulose | Same foods as listed for Sodium Carboxymethyl Cellulose | Same levels as prescribed for Sodium Carboxymethyl Cellulose |
| C.14 | Carob Bean Gum | (1) Cream; French dressing; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Relishes; Salad dressing | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Calorie-reduced margarine | (3) 0.5% in accordance with the requirements of paragraph B.09.017(b) | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Sour cream | (5) 0.5% in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (6) Unstandardized Foods | (6) Good Manufacturing Practice | ||
| (7) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (7) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| C.15 | Carrageenan | (1) Ale; Beer; Brawn; Canned (naming the poultry); Cream; French dressing; Headcheese; (naming the fruit) Jelly with pectin; Light beer; Malt liquor; Meat binder (when sold for use in prepared meat or prepared meat by-products in which a gelling agent is a permitted ingredient); Meat by-product loaf; Meat loaf; (naming the flavour) Milk; Mustard pickles; Porter; Potted meat; Potted meat by-product; Prepared fish or prepared meat (Division 21); Relishes; Salad dressing; (naming the flavour) Skim milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk with added milk solids; (naming the flavour) Partly skimmed milk with added milk solids; Stout | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Evaporated milk | (3) 0.015% | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Evaporated partly skimmed milk; Concentrated partly skimmed milk | (5) 0.01% | ||
| (6) Infant formula based on isolated amino acids or protein hydrolysates, or both | (6) 0.1% as consumed. If used in combination with algin or guar gum or both, the total not to exceed 0.1% | ||
| (7) Infant formula | (7) 0.03% as consumed. If used in combination with algin or guar gum or both, the total not to exceed 0.03% | ||
| (8) Unstandardized foods | (8) Good Manufacturing Practice | ||
| (9) Calorie-reduced margarine | (9) 0.5% in accordance with the requirements of section B.09.017 | ||
| (10) Sour cream | (10) 0.5%, in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (11) Canned asparagus; Canned green beans; Canned wax beans; Canned peas | (11) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| (12) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (12) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (13) Lactose-free infant formula based on milk protein | (13) 0.05% as consumed. If used in combination with algin or guar gum or both, the total not to exceed 0.05% | ||
| C.17 | Cellulose Gum | Same foods as listed for Sodium Carboxymethyl Cellulose | Same levels as prescribed for Sodium Carboxymethyl Cellulose |
| C.18 | Citric Acid Esters of Mono- and Di-glycerides | (1) Infant formula based on crystalline amino acids or protein hydrolysates, or both | (1) 0.155% as consumed |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| F.1 | Furcelleran | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Calorie-reduced margarine | (3) 0.5% in accordance with the requirements of section B.09.017 | ||
| (4) Canned asparagus; Canned green beans; Canned waxed beans; Canned peas | (4) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| G.1 | Gelatin | (1) Brawn; Canned (naming the poultry); Cream; Headcheese; Meat binder or (naming the meat product) binder where sold for use in prepared meat or prepared meat by-product in which a gelling agent is a permitted ingredient; Meat by-product loaf; Meat loaf; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jelly with pectin; Potted meat; Potted meat by-product; Prepared fish or prepared meat (Division 21); Prepared hams, shoulders, butts, picnics and backs; Relishes | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Sour cream | (4) 0.5% in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (5) Unstandardized Foods | (5) Good Manufacturing Practice | ||
| (6) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (6) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| G.2 | Gellan Gum | (1) Frostings; Unstandardized confectionery | (1) 0.5% |
| (2) Aspics; Unstandardized fruit spreads; Unstandardized processed fruit products | (2) 0.3% | ||
| (3) Calorie-reduced margarine; Reduced fat spreads | (3) 0.25% | ||
| (4) Unstandardized dairy products | (4) 0.15% | ||
| (5) Filling mixes; Fillings; French dressing; Pudding mixes; Puddings; Salad dressing; Unstandardized dressings; Unstandardized gelatins | (5) 0.1% | ||
| (6) Baking mixes; Unstandardized bakery products | (6) 0.1% of the dry mix | ||
| (7) Topping mixes; Toppings; Unstandardized sauces; Unstandardized table syrups | (7) 0.05% | ||
| (8) Unstandardized beverages | (8) 0.08% | ||
| (9) Snack foods | (9) 0.1% | ||
| G.3 | Guar Gum | (1) Bread; Cream; French dressing; Mincemeat; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Relishes; Salad dressing | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Infant formula | (3) 0.03% as consumed. If used in combination with algin or carrageenan or both, the total not to exceed 0.03% | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Calorie-reduced margarine | (6) 0.5% in accordance with the requirements of section B.09.017 | ||
| (7) Sour cream | (7) 0.5% in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (8) Canned asparagus; Canned green beans; Canned waxed beans; Canned peas | (8) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| (9) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (9) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (10) Infant formula based on isolated amino acids or protein hydrolysates, or both | (10) 0.1% as consumed. If used in combination with algin or carrageenan or both, the total not to exceed 0.1% | ||
| (11) Lactose-free infant formula based on milk protein | (11) 0.05% as consumed. If used in combination with algin or carrageenan or both, the total not to exceed 0.05% | ||
| G.4 | Gum Arabic | Same foods as listed for Acacia Gum | Same level as prescribed for Acacia Gum |
| H.1 | Hydroxylated Lecithin | (1) Chocolate products; Cocoa products | (1) 1.0% |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| H.1A | Hydroxypropyl Cellulose | Unstandardized foods | Good Manufacturing Practice |
| H.2 | Hydroxypropyl Methylcellulose | (1) French dressing; (naming the flavour) Milk; Mustard pickles; Relishes; (naming the flavour) Skim milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk with added milk solids; (naming the flavour) Partly skimmed milk with added milk solids; Salad dressing | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| I.1 | Irish Moss Gelose | Same foods as listed for Carrageenan | Same levels as prescribed for Carrageenan |
| K.1 | Karaya Gum | (1) French dressing; (naming the flavour) Milk; Mustard pickles; Relishes; (naming the flavour) Skim milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk with added milk solids; (naming the flavour) Partly skimmed milk with added milk solids; Salad dressing | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Calorie-reduced margarine | (5) 0.5% in accordance with the requirements of section B.09.017 | ||
| L.1 | Lactylated Mono- and Di-glycerides | (1) Shortening | (1) 8.0% (except that the total combined mono- and di-glycerides and lactylated mono- and di-glycerides must not exceed 20.0% of the shortening) |
| (2) Unstandardized foods | (2) 8.0% of the fat content | ||
| L.1A | Lactylic Esters of Fatty Acids | Unstandardized foods | Good Manufacturing Practice |
| L.2 | Lecithin | (1) Bread; Cream; (naming the flavour) Milk; Mustard pickles; Relishes; (naming the flavour) Skim milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk with added milk solids; (naming the flavour) Partly skimmed milk with added milk solids | (1) Good Manufacturing Practice |
| (2) Ice Cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5%, singly or in combination with other emulsifiers | ||
| (3) Infant formula | (3) 0.03% as consumed | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Margarine | (6) 0.2% | ||
| (7) Calorie-reduced margarine | (7) 0.5% | ||
| (8) Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (8) 0.2% | ||
| (9) Milk powder | (9) 0.5% | ||
| (10) Chocolate products; Cocoa products | (10) 1.0% | ||
| L.3 | Locust Bean Gum | Same foods as listed for Carob Bean Gum | Same levels as prescribed for Carob Bean Gum |
| M.1 | Magnesium Chloride | Tofu | 0.3%, calculated as the anhydrous salt |
| M.2 | Methylcellulose | (1) Ale; Beer; French dressing; Light beer; Malt liquor; Porter; Salad dressing; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| M.3 | Methyl Ethyl Cellulose | Unstandardized foods | Good Manufacturing Practice |
| M.4 | Mono-glycerides | (1) Bread; Cream; Fish paste | (1) Good Manufacturing Practice |
| (2) Chocolate products; Cocoa products | (2) 1.5% | ||
| (3) Ice cream mix; Ice milk mix | (3) A total of 0.5% of stabilizing agents in accordance with subparagraphs B.08.061(b)(vi) and B.08.071(b)(vi) | ||
| (4) Creamed cottage cheese | (4) Good Manufacturing Practice | ||
| (5) Infant formula | (5) 0.25% as consumed | ||
| (6) Sausage casings | (6) 0.35% of the casing | ||
| (7) Margarine | (7) 0.5% | ||
| (8) Sherbet | (8) 0.75% | ||
| (9) Shortening | (9) 10.0% (except that the total combined mono and diglycerides and lactylated mono and diglycerides must not exceed 20.0% of the shortening) | ||
| (10) Sour Cream | (10) 0.3% | ||
| (11) Unstandardized Foods | (11) Good Manufacturing Practice | ||
| (12) Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (12) 0.5% in accordance with the requirements of sections B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 | ||
| M.5 | Mono- and Di-glycerides | (1) Bread; Cream; Fish paste | (1) Good Manufacturing Practice |
| (2) Chocolate products; Cocoa products | (2) 1.5% | ||
| (3) Ice cream mix; Ice milk mix | (3) A total of 0.5% of stabilizing agents in accordance with subparagraphs B.08.061(b)(vi) and B.08.071(b)(vi) | ||
| (4) Cottage Cheese; Creamed Cottage Cheese | (4) Good Manufacturing Practice | ||
| (5) Infant formula | (5) 0.25% as consumed | ||
| (6) Sausage casings | (6) 0.35% of the casing | ||
| (7) Margarine | (7) 0.5% | ||
| (8) Sherbet | (8) 0.75% | ||
| (9) Shortening | (9) 10.0% (except that the total combined mono and diglycerides and lactylated mono and diglycerides must not exceed 20.0% of the shortening) | ||
| (10) Sour Cream | (10) 0.3% | ||
| (11) Unstandardized Foods | (11) Good Manufacturing Practice | ||
| (12) Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (12) 0.5% in accordance with the requirements of sections B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 | ||
| M.6 | Monosodium Salts of Phosphorylated Mono- and Diglycerides | (1) Edible vegetable oil-based cookware coating emulsions | (1) 4.0% |
| O.1 | Oat Gum | (1) Unstandardized Foods | (1) Good Manufacturing Practice |
| P.1 | Pectin | (1) Apple (or rhubarb) and (naming the fruit) Jam; Cream; Fig marmalade; Fig marmalade with pectin; French dressing; Mincemeat; Mustard pickles; (naming the citrus fruit) Marmalade with pectin; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; Pineapple marmalade; Pineapple marmalade with pectin; Relishes; Salad dressing | (1) Good Manufacturing Practice |
| (2) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sour cream | (3) 0.5% in accordance with the requirement of clause B.08.077(b)(vii)(A) | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| P.1A | Polyglycerol Esters of Fatty Acids | (1) Unstandardized foods | (1) Good Manufacturing Practice |
| (2) Vegetable oils | (2) 0.025% | ||
| (3) Calorie-reduced margarine | (3) 0.2% in accordance with the requirements of paragraph B.09.017(c) | ||
| P.1B | Polyglycerol Esters of Interesterified Castor Oil Fatty Acids | (1) Chocolate products | (1) 0.5% |
| (2) Unstandardized chocolate flavoured confectionery coatings | (2) 0.25% | ||
| (3) Edible vegetable oil-based pan coating emulsions for use on baking pans | (3) 2.0% | ||
| P.2 | Polyoxyethylene (20) Sorbitan Monooleate; Polysorbate 80 | (1) Ice cream; Ice cream mix; Ice milk; Ice milk mix; Sherbet | (1) 0.1%. If Polyoxyethylene (20) sorbitan tristearate is also used, the total must not exceed 0.1% |
| (2) Unstandardized frozen desserts | (2) 0.1% | ||
| (3) Pickles and relishes | (3) 0.05% | ||
| (4) Unstandardized beverage bases; Unstandardized beverage mixes | (4) 0.05% of the beverage. If sorbitan monostearate is also used the total must not exceed 0.05% of the beverage | ||
| (5) Imitation dry cream mix | (5) 0.1%. If Polyoxyethylene (20) sorbitan monostearate, Polyoxyethylene (20) sorbitan tristearate or Sorbitan monostearate, either singly or in combination is also used, the total must not exceed 0.4% | ||
| (6) Whipped vegetable oil topping | (6) 0.05%. If Polyoxyethylene (20) sorbitan monostearate, Polyoxyethylene (20) sorbitan tristearate or Sorbitan monostearate, either singly or in combination is also used, the total must not exceed 0.4% | ||
| (7) Cake icing; cake icing mix | (7) 0.5% of the finished cake icing. If Polyoxyethylene (20) sorbitan monostearate, or Sorbitan monostearate, either singly or in combination is also used, the total must not exceed 0.5% of the finished cake icing | ||
| (8) Salt | (8) 10 p.p.m. | ||
| (9) Whipped cream | (9) 0.1% | ||
| (10) Breath freshener products | (10) 100 p.p.m. | ||
| (11) Creamed cottage cheese | (11) 80 p.p.m. | ||
| (12) Spice oils and spice oleoresins for use in pumping pickle employed in the curing of preserved meat or preserved meat by-product (Division 14) | (12) 0.2% of the pumping pickle | ||
| (13) Sausage casings | (13) 0.15% of the casing | ||
| (14) Liquid Smoke Flavours | (14) Good Manufacturing Practice. Residues of Polysorbate 80 must not exceed 275 p.p.m. in the finished food | ||
| (15) Vegetable oils | (15) 0.125% | ||
| (16) Annatto formulations | (16) 25% of the total colour formulation | ||
| (17) Turmeric formulations | (17) 50% of the total colour formulation | ||
| (18) Liquid smoke flavour concentrate | (18) Good Manufacturing Practice. Residues of Polysorbate 80 must not exceed 0.3% in the finished food. | ||
| (19) Unstandardized salad dressings | (19) 0.25% | ||
| P.3 | Polyoxyethyl ene (20) Sorbitan Monostearate; Polysorbate 60 | (1) Imitation dry cream mix; Vegetable oil creaming agent; Whipped vegetable oil topping; Vegetable oil topping mix | (1) 0.4%. If Polyoxyethylene (20) sorbitan tristearate, Sorbitan monostearate or Polyoxyethylene (20) sorbitan mono-oleate, either singly or in combination is also used, the total must not exceed 0.4%, except that in the case of whipped vegetable oil topping a combination of Polysorbate 60 and Sorbitan monostearate may be used in excess of 0.4%, if the amount of the Polysorbate 60 does not exceed 0.77% and the amount of Sorbitan monostearate does not exceed 0.27% of the whipped vegetable oil topping |
| (2) Cakes | (2) 0.5% on a dry weight basis. If Polyoxyethylene (20) sorbitan tristearate is also used, the total must not exceed 0.5% on a dry weight basis | ||
| (3) Cakes; Cake mixes | (3) 0.5% on a dry weight basis. If Sorbitan monostearate is also used, the total must not exceed 0.7% on a dry weight basis | ||
| (4) Unstandardized confectionery coatings and unstandardized moulded confectionery products for use as confectionery or in baking | (4) 0.5%. If any combination of Polyoxyethylene (20) Sorbitan tristearate, Sorbitan monostearate or Sorbitan tristearate are all used the total must not exceed 1.0% | ||
| (5) Cake icing; Cake icing mix | (5) 0.5% of the finished cake icing. If Sorbitan monostearate or Polyoxyethylene (20) sorbitan mono-oleate either singly or in combination is also used, the total must not exceed 0.5% of the finished cake icing | ||
| (6) Pie fillings; Puddings | (6) 0.5% on a dry weight basis | ||
| (7) Unstandardized beverage bases; Unstandardized beverage mixes | (7) 0.05% of the beverage. If Sorbitan monostearate is also used the total must not exceed 0.05% of the beverage | ||
| (8) Sour Cream Substitute | (8) 0.1% | ||
| (9) Unstandardized dressings; Unstandardized prepared canned cooking sauces | (9) 0.3% | ||
| (10) Fat base formulation for self-basting of poultry by injection | (10) 0.25% | ||
| (11) Unstandardized sandwich spreads; Unstandardized dips | (11) 0.2% | ||
| (12) Dry soup base or mix | (12) 250 p.p.m. in soup as consumed | ||
| (13) Dry batter coating mixes | (13) 0.5% of the dry mix | ||
| (14) Prepared alcoholic cocktails | (14) 120 p.p.m. in beverage as consumed | ||
| P.4 | Polyoxyethylene (20) Sorbitan Tristearate; Polysorbate 65 | (1) (Naming the flavour) Milk; (naming the flavour) Skim milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk with added milk solids; (naming the flavour) Partly skimmed milk with added milk solids | (1) 0.5% |
| (2) Ice cream; Ice cream mix; Ice milk; Ice milk mix; Sherbet | (2) 0.1%. If Polyoxyethylene (20) sorbitan mono-oleate is also used, the total must not exceed 0.1% | ||
| (3) Unstandardized frozen desserts | (3) 0.1% | ||
| (4) Cakes | (4) 0.3% on a dry weight basis. If Polyoxyethylene (20) sorbitan monostearate is also used, the total must not exceed 0.5% on a dry weight basis | ||
| (5) Unstandardized confectionery coatings | (5) 0.5%. If any combination of Polyoxyethylene (20) sorbitan monostearate, Sorbitan monostearate, or Sorbitan tristearate are also used, the total must not exceed 1.0% | ||
| (6) Unstandardized beverage bases; Unstandardized beverage mixes | (6) 0.05% of the beverage. If Sorbitan monostearate is also used, the total must not exceed 0.05% of the beverage | ||
| (7) Imitation dry cream mix; Vegetable oil creaming agent; Whipped vegetable oil topping; Vegetable oil topping mix | (7) 0.4%. If Polyoxyethylene (20) sorbitan monostearate, Sorbitan monostearate or Polyoxyethylene (20) sorbitan mono-oleate, either singly or in combination is also used, the total must not exceed 0.4% | ||
| (8) Breath freshener products | (8) 200 p.p.m. | ||
| P.5 | Polyoxyethylene (8) Stearate | Unstandardized bakery products | 0.4% |
| P.6 | Potassium Alginate | Same foods as listed for Algin | Same levels as prescribed for Algin |
| P.7 | Potassium Carrageenan | Same foods as listed for Carrageenan | Same levels as prescribed for Carrageenan |
| P.8 | Potassium Chloride | Unstandardized foods | Good Manufacturing Practice |
| P.9 | Potassium Citrate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| P.10 | Potassium Furcelleran | Same foods as listed for Furcelleran | Same levels as prescribed for Furcelleran |
| P.11 | Potassium Phosphate, dibasic | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| P.12 | Propylene Glycol Alginate | (1) Ale; Beer; French dressing; Light beer; Malt liquor; Mustard pickles; Porter; Relishes; Salad dressing; Stout | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Calorie-reduced margarine | (5) 0.5% in accordance with the requirements of section B.09.017 | ||
| (6) Sour cream | (6) 0.5% in accordance with the requirements of clause B.08.077(b)(vii)(A) | ||
| (7) Canned asparagus; Canned green beans; Canned wax beans; Canned peas | (7) 1.0% in accordance with the requirements of clause B.11.002(d)(viii)(C) | ||
| (8) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (8) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| P.13 | Propylene Glycol Ether of Methylcellulose | Same foods as listed for Hydroxypropyl Methylcellulose | Same levels as prescribed for Hydroxypropyl Methylcellulose |
| P.14 | Propylene Glycol Mono Fatty Acid Esters | (1) Ice cream mix | (1) 0.35% of the ice cream made from the mix |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.1 | Sodium Acid Pyrophosphate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| S.2 | Sodium Alginate | (1) Same foods as listed for Algin | (1) Same levels as prescribed for Algin |
| (2) Coarse crystal salt | (2) 15 p.p.m. | ||
| (3) Glaze of frozen fish | (3) Good Manufacturing Practice | ||
| S.2A | Sodium Aluminum Phosphate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| S.3 | Sodium Carboxymethyl Cellulose | (1) Cream; French dressing; Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Relishes; Salad dressing | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Glaze of frozen fish | (5) Good Manufacturing Practice | ||
| (6) Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients) | (6) 0.5% | ||
| (7) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (7) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| S.4 | Sodium Carrageenan | Same foods as listed for Carrageenan | Same levels as prescribed for Carrageenan |
| S.5 | Sodium Cellulose Glycolate | Same foods as listed for Sodium Carboxymethyl Cellulose | Same levels as prescribed for Sodium Carboxymethyl Cellulose |
| S.6 | Sodium Citrate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Evaporated milk; evaporated skim milk or concentrated skim milk; evaporated partly skimmed milk or concentrated partly skimmed milk | (2) 0.1% singly or in combination with sodium phosphate, dibasic | ||
| (3) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (3) 0.5% | ||
| (4) Sherbet | (4) 0.75% | ||
| S.7 | Sodium Furcelleran | Same foods as listed for Furcelleran | Same levels as prescribed for Furcelleran |
| S.8 | Sodium Gluconate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| S.9 | Sodium Hexameta-phosphate | (1) Mustard pickles; Relishes | (1) Good Manufacturing Practice |
| (2) Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Infant formula | (3) 0.05% as consumed | ||
| (4) Sherbet | (4) 0.75% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (6) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 | ||
| (7) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (7) 0.1% | ||
| S.11 | Sodium Phosphate, dibasic | (1) Mustard pickles; (naming the flavour) Milk; (naming the flavour) Partly skimmed milk; (naming the flavour) Partly skimmed milk with added milk solids; (naming the flavour) Skim milk; (naming the flavour) Skim milk with added milk solids; Relishes | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese | (2) 0.5% | ||
| (3) Evaporated milk; evaporated skim milk or concentrated skim milk; evaporated partly skimmed milk or concentrated partly skimmed milk | (3) 0.1% singly or in combination with sodium citrate | ||
| (4) Sour cream | (4) 0.05% in accordance with the requirements of clause B.08.077(b)(vii)(C) | ||
| (5) Unstandardized Foods | (5) Good Manufacturing Practice | ||
| (6) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (6) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 | ||
| S.12 | Sodium Phosphate, monobasic | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.13 | Sodium Phosphate, tribasic | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.14 | Sodium Potassium Tartrate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.15 | Sodium Pyrophosphate, tetrabasic | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 3.5%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (3) 0.1% | ||
| S.15A | Sodium Stearoyl-2-Lac-tylate | (1) Icing and icing mixes | (1) 0.4% of dry ingredient weight |
| (2) Fillings and filling mixes | (2) 0.5% of dry ingredient weight | ||
| (3) Puddings and pudding mixes | (3) 0.2% of the finished product | ||
| (4) Sour cream substitutes | (4) 1.0% of dry ingredient weight | ||
| (5) Vegetable oil creaming agents | (5) 2.0% of dry ingredient weight | ||
| (6) Batter mix | (6) 0.75% of dry ingredient weight | ||
| (7) Unstandardized cream-based liquors | (7) 0.35% of the finished product | ||
| (8) Salad dressing; French dressing | (8) 0.4% of the finished product | ||
| (9) Soups | (9) 0.2% of the finished product | ||
| S.16 | Sodium Tartrate | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (1) 4.0%, in accordance with the requirements of sections B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3 and B.08.041.4 |
| S.16A | Sodium Tripolyphosphate | A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | 0.1% |
| S.18 | Sorbitan Monostearate | (1) Imitation dry cream mix; Vegetable oil creaming agent; Whipped vegetable oil topping; Vegetable oil topping mix | (1) 0.4%. If Polyoxyethylene (20) sorbitan tristearate, Polysorbate 60 or Polyoxyethylene (20) sorbitan mono-oleate, either singly or in combination is also used, the total must not exceed 0.4%, except that in the case of whipped vegetable oil topping a combination of Sorbitan monostearate and Polysorbate 60 may be used in excess of 0.4% if the amount of Sorbitan monostearate does not exceed 0.27% and the amount of Polysorbate 60 does not exceed 0.77% of the weight of the whipped vegetable oil topping |
| (2) Cake; Cake mix | (2) 0.6% on a dry weight basis. If Polyoxyethylene (20) sorbitan monostearate is also used, the total must not exceed 0.7% on a dry weight basis | ||
| (3) Unstandardized confectionery coatings and unstandardized moulded confectionery products for use as confectionery or in baking | (3) 1.0%. If any combination of Polyoxyethylene (20) sorbitan monostearate, Polyoxyethylene (20) sorbitan tristrearate or Sorbitan tristearate are also used, the total must not exceed 1.0% | ||
| (4) Cake icing; Cake icing mix | (4) 0.5% of the finished cake icing. If Polyoxyethylene (20) sorbitan mono-oleate or Polyoxyethylene (20) sorbitan monostearate, either singly or in combination is also used, the total must not exceed 0.5% of the finished cake icing | ||
| (5) Unstandardized beverage bases; Unstandardized beverage mixes | (5) 0.05% of the beverage. If Polyoxyethylene (20) sorbitan mono-oleate is also used, the total must not exceed 0.05% of the beverage. If Polyoxyethylene (20) sorbitan monostearate is also used, the total must not exceed 0.05% of the beverage. If Polyoxyethylene (20) sorbitan tristearate is also used, the total must not exceed 0.05% of the beverage | ||
| (6) Dry soup base or mix | (6) 250 p.p.m. in soup as consumed | ||
| (7) Dried yeast | (7) 1.5% (Residues of sorbitan monostearate in bread and other yeast leavened bakery products not to exceed 0.05%). | ||
| (8) Chocolate products | (8) 1.0% | ||
| (9) Puddings | (9) 0.5% | ||
| S.18A | Sorbitan trioleate | Sausage casings | 0.35% of the casing |
| S.18B | Sorbitan Tristearate | (1) Margarine; Shortening | (1) 1.0% |
| (2) Unstandardized confectionery coatings and unstandardized moulded confectionery products for use as a confectionery or in baking | (2) 1.0% If any combination of Polyoxyethylene (20) sorbitan monostearate, Polyoxyethylene (20) sorbitan tristearate or Sorbitan monostearate are also used, the total must not exceed 1.0% | ||
| (3) Ice cream mix | (3) 0.035% of the ice cream made from the mix | ||
| (4) Unstandardized frozen desserts | (4) 0.035% | ||
| S.19 | Stearyl Monoglyceridyl Citrate | Shortening | Good Manufacturing Practice |
| S.20 | Sucrose esters of fatty acids | (1) Carotenoid colour preparations | (1) 1.5% |
| (2) Unstandardized confectionery; Unstandardized confectionery coatings | (2) 0.5% | ||
| T.2 | [Repealed, SOR/2006-91, s. 5] | ||
| T.3 | Tragacanth Gum | (1) French dressing; Mustard pickles; Salad dressing; Relishes | (1) Good Manufacturing Practice |
| (2) Cottage cheese; Creamed cottage cheese; Ice cream; Ice cream mix; Ice milk; Ice milk mix | (2) 0.5% | ||
| (3) Sherbet | (3) 0.75% | ||
| (4) Lumpfish Caviar | (4) 1.0% | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Calorie-reduced margarine | (6) 0.5% in accordance with the requirements of section B.09.017 | ||
| (7) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (7) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (8) Comminuted prepared fish or prepared meat, other than lumpfish caviar; Comminuted preserved fish or preserved meat (Division 21) | (8) 0.75% | ||
| X.1 | Xanthan Gum | (1) French Dressing; Salad Dressing; Unstandardized Foods | (1) Good Manufacturing Practice |
| (2) Cottage Cheese; Creamed Cottage Cheese | (2) 0.5% or, if used in combination with other stabilizing agents, the total amount of the combined stabilizers shall not exceed 0.5% | ||
| (3) Calorie-reduced margarine | (3) 0.5% in accordance with the requirements of section B.09.017 | ||
| (4) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (4) 0.5%, in accordance with the requirements of sections B.08.035, B.08.037, B.08.038, B.08.039, B.08.041.3, B.08.041.4, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (5) Mustard pickles; relishes | (5) 0.1% | ||
| (6) Ice Cream Mix | (6) 0.1% or, if used in combination with microcrystalline cellulose and other stabilizers, the total amount of combined stabilizers and microcrystalline cellulose shall not exceed 0.5% | ||
| (7) Ice Milk Mix | (7) 0.1% or, if used in combination with other stabilizers, the total amount of combined stabilizers shall not exceed 0.5% | ||
| (8) Sherbet | (8) 0.1% or, if used in combination with other stabilizers, the total amount of combined stabilizers shall not exceed 0.75% | ||
| (9) Cream for whipping, heat-treated above 100°C | (9) 0.02% | ||
- SOR/78-403, ss. 5(F) to 13(F), 14 to 16, 17(F) to 21(F), 22
- SOR/78-656, ss. 14, 15
- SOR/78-876, s. 2
- SOR/79-660, ss. 5 to 10
- SOR/79-752, s. 6
- SOR/80-501, s. 4
- SOR/81-60, ss. 7 to 10
- SOR/81-565, ss. 4, 5
- SOR/81-934, ss. 2 to 6
- SOR/82-383, s. 9
- SOR/82-1071, ss. 9 to 16
- SOR/83-932, ss. 3, 4
- SOR/84-300, s. 50(E)
- SOR/84-602, s. 2
- SOR/84-801, s. 3
- SOR/85-179, ss. 2 to 4
- SOR/85-623, s. 3(E)
- SOR/88-99, s. 3
- SOR/88-419, ss. 2, 3
- SOR/90-87, s. 9
- SOR/91-710, s. 1
- SOR/92-64, s. 1
- SOR/92-93, s. 2
- SOR/92-344, s. 1
- SOR/93-466, ss. 3, 4
- SOR/94-38, s. 2
- SOR/94-567, s. 2
- SOR/94-689, s. 2(F)
- SOR/96-160, s. 2
- SOR/96-376, s. 1
- SOR/96-497, s. 1
- SOR/96-499, s. 1
- SOR/97-29, s. 1
- SOR/97-263, ss. 4 to 10
- SOR/98-580, s. 1(F)
- SOR/2000-353, s. 7(F)
- SOR/2005-316, s. 1
- SOR/2005-395, ss. 1 to 4, 5(F)
- SOR/2006-91, ss. 4, 5
- SOR/2007-75, ss. 4 to 6
- SOR/2007-76, ss. 1, 2
- SOR/2010-41, s. 9(E)
- SOR/2010-94, ss. 8(E), 9(E)
- SOR/2010-142, ss. 9, 10(F), 11 to 13, 14(F), 15(F), 16
- SOR/2011-235, ss. 3 to 6, 16(F)
- SOR/2011-278, ss. 11, 12(E)
- SOR/2012-43, ss. 21 to 25, 26(F), 27, 28
- SOR/2012-104, ss. 3, 4(F), 5 to 9, 20(F)
- SOR/2012-106, s. 1
TABLE V
Food Additives That May Be Used as Food Enzymes
| Item No. | Column I | Column II | Column III | Column IV |
|---|---|---|---|---|
| Additive | Permitted Source | Permitted in or Upon | Maximum Level of Use | |
| A.01 | α-Acetolactate decarboxylase | Bacillus subtilis ToC46 (pUW235) | (1) Brewers’ Mash | (1) Good Manufacturing Practice |
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| A.02 | Aminopeptidase | Lactococcus lactis | (1) Cheddar cheese; (naming the variety) Cheese | (1) Good Manufacturing Practice |
| (2) Dairy based flavouring preparations | (2) Good Manufacturing Practice | |||
| (3) Hydrolyzed animal, milk and vegetable protein | (3) Good Manufacturing Practice | |||
| A.1 | Amylase | Aspergillus niger var.; Aspergillus oryzae var.; Bacillus subtilis var.; Rhizopus oryzae var.; Barley Malt | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bread; Flour; Whole wheat flour | (2) Good Manufacturing Practice | |||
| (3) Cider; Wine | (3) Good Manufacturing Practice | |||
| (4) Chocolate syrups | (4) Good Manufacturing Practice | |||
| (5) Distillers’ Mash | (5) Good Manufacturing Practice | |||
| (6) Malt-flavoured dried breakfast cereals | (6) Good Manufacturing Practice | |||
| (7) Single-strength fruit juices | (7) Good Manufacturing Practice | |||
| (8) Pre-cooked (instant) breakfast cereals | (8) Good Manufacturing Practice | |||
| (9) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup) | (9) Good Manufacturing Practice | |||
| (10) Unstandardized bakery products | (10) Good Manufacturing Practice | |||
| (11) Plant-based beverages | (11) Good Manufacturing Practice | |||
| (12) Infant cereal products | (12) Good Manufacturing Practice | |||
| Aspergillus niger STz18-9 (pHUda7) | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Starch used in the production of dextrins, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup), maltose | (3) Good Manufacturing Practice | |||
| Bacillus amyloliquefaciens EBA 20 (pUBH2); Bacillus licheniformis; Bacillus licheniformis BML 592 (pAmyAmp); Bacillus licheniformis BML 730 (pAmyAmp); Bacillus licheniformis LA 57 (pDN1981); Bacillus licheniformis LAT8 (pLAT3); Bacillus licheniformis LiH 1159 (pLiH1108); Bacillus licheniformis LiH 1464 (pLiH1346); Bacillus licheniformis PL 1303 (pPL1117); Bacillus licheniformis MOL2083 (pCA164-LE399) | (1) Distillers’ Mash | (1) Good Manufacturing Practice | ||
| (2) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup) | (2) Good Manufacturing Practice | |||
| (3) Brewers’ mash | (3) Good Manufacturing Practice | |||
| Bacillus licheniformis 3253 (plCatH-3253); Bacillus licheniformis 3266 (plCatH-3266ori1); Bacillus stearothermophilus; Bacillus subtilis B1.109 (pCPC800) | (1) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup) | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Brewers’ Mash | (3) Good Manufacturing Practice | |||
| (4) Bread; Flour; Whole wheat flour | (4) Good Manufacturing Practice | |||
| (5) Unstandardized bakery products | (5) Good Manufacturing Practice | |||
| Bacillus subtilis B1.109 (pCPC720) (ATCC 39, 705) | (1) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup) | (1) Good Manufacturing Practice | ||
| A.2 | Amylase (maltogenic) | Bacillus subtilis BRG-1 (pBRG1); Bacillus subtilis DN1413 (pDN1413); Bacillus subtilis LFA 63 (pLFA63); Bacillus subtilis RB-147 (pRB147) | (1) Starch used in the production of dextrins, maltose, dextrose, glucose, (glucose syrup) or glucose solids (dried glucose syrup) | (1) Good manufacturing practice |
| (2) Bread; Flour; Whole wheat flour | (2) Good manufacturing practice | |||
| (3) Unstandardized bakery products | (3) Good manufacturing practice | |||
| A.3 | Asparaginase | Aspergillus niger ASP72; Aspergillus oryzae (pCaHj621/BECh2#10) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | |||
| B.1 | Bovine Rennet | Aqueous extracts from the fourth stomach of adult bovine animals, sheep and goats | Cheddar cheese; Cottage cheese; Cream cheese; Cream cheese spread; Cream cheese spread with (naming the added ingredients); Cream cheese with (naming the added ingredients); (naming the variety) Cheese | Good Manufacturing Practice |
| B.2 | Bromelain | The pineapples Ananas comosus and Ananas bracteatus | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bread; Flour; Whole wheat flour | (2) Good Manufacturing Practice | |||
| (3) Sausage casings | (3) Good Manufacturing Practice | |||
| (4) Hydrolyzed animal, milk and vegetable protein | (4) Good Manufacturing Practice | |||
| (5) Meat cuts | (5) Good Manufacturing Practice | |||
| (6) Meat tenderizing preparations | (6) Good Manufacturing Practice | |||
| (7) Pumping pickle for the curing of beef cuts | (7) Good Manufacturing Practice in accordance with paragraph B.14.009(g) | |||
| (8) Sugar wafers, waffles, pancakes | (8) Good Manufacturing Practice | |||
| C.1 | Catalase | Aspergillus niger var.; Micrococcus lysodeikticus; Bovine (Bos taurus) liver | (1) Soft drinks | (1) Good Manufacturing Practice |
| (2) Liquid egg-white (liquid albumen), liquid whole egg or liquid yolk, destined for drying | (2) Good Manufacturing Practice | |||
| (3) Liquid whey treated with hydrogen peroxide in accordance with item H.1, Table VIII | (3) Good Manufacturing Practice | |||
| C.2 | Cellulase | Aspergillus niger var. | (1) Distillers’ Mash | (1) Good Manufacturing Practice |
| (2) Liquid coffee concentrate | (2) Good Manufacturing Practice | |||
| (3) Spice extracts; Natural flavour and colour extractives | (3) Good Manufacturing Practice | |||
| Trichoderma reesei QM 9414 | (1) Single-strength fruit juices | (1) Good Manufacturing Practice | ||
| (2) Tea leaves for the production of tea solids | (2) Good Manufacturing Practice | |||
| C.3 | Chymosin | |||
| (i) Chymosin A | Escherichia coli K-12, GE81 (pPFZ87A) | (1) Cheddar cheese; (naming the variety) cheese; Cottage cheese; Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Sour cream | (1) Good Manufacturing Practice | |
| (2) Unstandardized milk-based dessert preparations | (2) Good Manufacturing Practice | |||
| (ii) Chymosin B | Aspergillus niger var. awamori, GCC0349 (pGAMpR); Kluyveromyces marxianus var. lactis, DS1182 (pKS105) | (1) Cheddar cheese; (naming the variety) cheese; Cottage cheese; Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Sour cream | (1) Good Manufacturing Practice | |
| (2) Unstandardized milk-based dessert preparations | (2) Good Manufacturing Practice | |||
| F.1 | Ficin | Latex of fig tree (Ficus sp.) | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Sausage casings | (2) Good Manufacturing Practice | |||
| (3) Hydrolyzed animal, milk and vegetable protein | (3) Good Manufacturing Practice | |||
| (4) Meat cuts | (4) Good Manufacturing Practice | |||
| (5) Meat tenderizing preparations | (5) Good Manufacturing Practice | |||
| (6) Pumping pickle for the curing of beef cuts | (6) Good Manufacturing Practice in accordance with paragraph B.14.009(g) | |||
| G.1 | Glucoamylase (Amyloglucosidase; Maltase) | Aspergillus niger var.; Aspergillus oryzae var.; Rhizopus oryzae var. | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bread; Flour; Whole wheat flour | (2) Good Manufacturing Practice | |||
| (3) Chocolate syrups | (3) Good Manufacturing Practice | |||
| (4) Distillers’ Mash | (4) Good Manufacturing Practice | |||
| (5) Pre-cooked (instant) breakfast cereals | (5) Good Manufacturing Practice | |||
| (6) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup), or glucose solids (dried glucose syrup) | (6) Good Manufacturing Practice | |||
| (7) Unstandardized bakery products | (7) Good Manufacturing Practice | |||
| Aspergillus niger STz18-9 (pHUda7) | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Starch used in the production of dextrins, dextrose, glucose (glucose syrup) or glucose solids (dried glucose syrup), maltose | (3) Good Manufacturing Practice | |||
| Rhizopus niveus var. | (1) Distillers’ Mash | (1) Good Manufacturing Practice | ||
| (2) Mash destined for vinegar manufacture | (2) Good Manufacturing Practice | |||
| Rhizopus delemar var.; Multiplici sporus | (1) Brewers’ Mash | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Mash destined for vinegar manufacture | (3) Good Manufacturing Practice | |||
| (4) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup), or glucose solids (dried glucose syrup) | (4) Good Manufacturing Practice | |||
| G.2 | Glucanase | Aspergillus niger var.; Bacillus subtilis var. | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Corn for degermination | (2) Good Manufacturing Practice | |||
| (3) Distillers’ Mash | (3) Good Manufacturing Practice | |||
| (4) Mash destined for vinegar manufacture | (4) Good Manufacturing Practice | |||
| (5) Unstandardized bakery products | (5) Good Manufacturing Practice | |||
| Humicola insolens var. | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| G.3 | Glucose oxidase | Aspergillus niger var.; Aspergillus oryzae Mtl-72 (pHUda107) | (1) Soft drinks | (1) Good Manufacturing Practice |
| (2) Liquid egg-white (liquid albumen), liquid whole egg or liquid yolk, destined for drying | (2) Good Manufacturing Practice in accordance with paragraphs B.22.034(b), B.22.035(b) and B.22.036(b) | |||
| (3) Bread; flour; Whole wheat flour | (3) Good manufacturing practice | |||
| (4) Unstandardized bakery products | (4) Good manufacturing practice | |||
| G.4 | Glucose Isomerase | Bacillus coagulans var.; Streptomyces olivochromogenes var.; Actinoplanes missouriensis var.; Streptomyces olivaceus var.; Microbacterium arborescens NRRL B-11022; Streptomyces murinus DSM 3252; Streptomyces rubiginosus ATCC No. 21,175; Streptomyces rubiginosus SYC 5406 (pSYC5239) | (1) Glucose (glucose syrup) to be partially or completely isomerized to fructose | (1) Good Manufacturing Practice |
| H.1 | Hemicellulase | Bacillus subtilis var. | (1) Distillers’ Mash | (1) Good Manufacturing Practice |
| (2) Liquid coffee concentrate | (2) Good Manufacturing Practice | |||
| (3) Mash destined for vinegar manufacture | (3) Good Manufacturing Practice | |||
| I.01 | Inulinase | Aspergillus niger var. Tieghem | Inulin | Good Manufacturing Practice |
| I.1 | Invertase | Aspergillus japonicus | Sucrose used in the production of fructooligosaccharides | Good Manufacturing Practice |
| Saccharomyces sp. | (1) Unstandardized soft-centred and liquid-centred confectionery | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| L.1 | Lactase | Aspergillus niger var.; Aspergillus oryzae var.; Kluyveromyces fragilis (Kluyveromyces marxianus var. marxianus); Kluyveromyces lactis (Kluyveromyces marxianus var. lactis); Saccharomyces sp. | (1) Lactose-reducing enzyme preparations | (1) Good Manufacturing Practice |
| (2) Milk destined for use in ice cream mix | (2) Good Manufacturing Practice | |||
| (3) Bread; Flour; whole wheat flour | (3) Good Manufacturing Practice | |||
| (4) (naming the flavour) milk; (naming the flavour) skim milk; (naming the flavour) partly skimmed milk; (naming the flavour) malted milk; (naming the flavour) skimmed milk with added milk solids; (naming the flavour) partly skimmed milk with added milk solids | (4) Good Manufacturing Practice | |||
| Cell-free extracts from Candida pseudotropicalis | (1) Milk destined for use in ice cream mix | (1) Good Manufacturing Practice | ||
| (2) Yogurt | (2) Good Manufacturing Practice | |||
| (3) Whey | (3) Good Manufacturing Practice | |||
| (4) (naming the flavour) milk; (naming the flavour) skim milk; (naming the flavour) partly skimmed milk; (naming the flavour) malted milk; (naming the flavour) skim milk with added milk solids; (naming the flavour) partly skimmed milk with added milk solids | (4) Good Manufacturing Practice | |||
| L.2 | Lipase | Animal pancreatic tissue; Aspergillus niger var.; Aspergillus oryzae var.; Edible forestomach tissue of calves, kids or lambs; Rhizopus oryzae var. | (1) Dairy based flavouring preparations | (1) Good Manufacturing Practice |
| (2) Dried egg-white (dried albumen); Liquid egg-white ( liquid albumen) | (2) Good Manufacturing Practice | |||
| (3) Cheddar cheese; (naming the variety) Cheese; Processed cheddar cheese | (3) Good Manufacturing Practice | |||
| (4) Bread; Flour; Whole wheat flour | (4) Good Manufacturing Practice | |||
| (5) Unstandardized bakery products | (5) Good Manufacturing Practice | |||
| (6) Hydrolyzed animal, milk and vegetable protein | (6) Good Manufacturing Practice | |||
| Aspergillus oryzae (MLT-2) (pRML 787) (p3SR2); Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)); Rhizopus niveus | (1) Modified fats and oils | (1) Good Manufacturing Practice | ||
| (2) Cheddar cheese; (naming the variety) Cheese | (2) Good Manufacturing Practice | |||
| (3) Dairy based flavouring preparations | (3) Good Manufacturing Practice | |||
| (4) Hydrolyzed animal, milk and vegetable protein | (4) Good Manufacturing Practice | |||
| Aspergillus oryzae AI-11 (pBoel 960) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| (3) Modified fats and oils | (3) Good Manufacturing Practice | |||
| Aspergillus oryzae BECh2#3 (pCaHj559); Aspergillus oryzae (MStr115) (pMStr20) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| (3) Modified lecithin | (3) Good Manufacturing Practice | |||
| (4) Unstandardized egg products | (4) Good Manufacturing Practice | |||
| Aspergillus niger (pCaHj600/MBin118#11) | Modified fats and oils | Good Manufacturing Practice | ||
| Penicillium camembertii | (1) Edible fats and oils | (1) Good Manufacturing Practice | ||
| L.3 | Lipoxidase | Soyabean whey or meal | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice |
| L.4 | Lysozyme | Egg-white | Cheddar cheese; (naming the variety) Cheese | Good Manufacturing Practice |
| M.1 | Milk coagulating enzyme | Rhizomucor miehei (Cooney and Emerson) (previous name: Mucor miehei (Cooney and Emerson)) or Mucor pusillus Lindt by pure culture fermentation process or Aspergillus oryzae RET-1 (pBoel777) | (1) Cheddar cheese; Cottage cheese; (naming the variety) Cheese; Sour cream | (1) Good Manufacturing Practice |
| (2) Dairy based flavouring preparations | (2) Good Manufacturing Practice | |||
| (3) Hydrolyzed animal, milk and vegetable protein | (3) Good Manufacturing Practice | |||
| Endothia parasitica by pure culture fermentation processes | (1) Emmentaler (Emmental, Swiss) Cheese | (1) Good Manufacturing Practice | ||
| (2) Parmesan Cheese | (2) Good Manufacturing Practice | |||
| (3) Romano Cheese | (3) Good Manufacturing Practice | |||
| (4) Mozzarella (Scamorza) Cheese | (4) Good Manufacturing Practice | |||
| (5) Part Skim Mozzarella (Part Skim Scamorza) Cheese | (5) Good Manufacturing Practice | |||
| P.1 | Pancreatin | Pancreas of the hog (Sus scrofa) or ox (Bos taurus) | (1) Dried egg-white (dried albumen); Liquid egg-white (liquid albumen) | (1) Good Manufacturing Practice |
| (2) Pre-cooked (instant) breakfast cereals | (2) Good Manufacturing Practice | |||
| (3) Starch used in the production of dextrins, maltose, dextrose, glucose (glucose syrup), or glucose solids (dried glucose syrup) | (3) Good Manufacturing Practice | |||
| (4) Hydrolyzed animal, milk and vegetable proteins | (4) Good Manufacturing Practice | |||
| P.2 | Papain | Fruit of the papaya Carica papaya L. (Fam. Caricaceae) | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Beef before slaughter | (2) Good Manufacturing Practice | |||
| (3) Sausage casings; Water-soluble edible collagen films | (3) Good Manufacturing Practice | |||
| (4) Hydrolyzed animal, milk and vegetable protein | (4) Good Manufacturing Practice | |||
| (5) Meat cuts | (5) Good Manufacturing Practice | |||
| (6) Meat tenderizing preparations | (6) Good Manufacturing Practice | |||
| (7) Pre-cooked (instant) breakfast cereals | (7) Good Manufacturing Practice | |||
| (8) Pumping pickle for the curing of beef cuts | (8) Good Manufacturing Practice | |||
| (9) Unstandardized bakery products | (9) Good Manufacturing Practice | |||
| P.3 | Pectinase | Aspergillus niger var.; Rhizopus oryzae var. | (1) Cider; Wine | (1) Good Manufacturing Practice |
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Single-strength fruit juices | (3) Good Manufacturing Practice | |||
| (4) Natural flavour and colour extractives | (4) Good Manufacturing Practice | |||
| (5) Skins of citrus fruits destined for jam, marmalade and candied fruit production | (5) Good Manufacturing Practice | |||
| (6) Vegetable stock for use in soups | (6) Good Manufacturing Practice | |||
| (7) Tea leaves for the production of tea solids | (7) Good Manufacturing Practice | |||
| Aspergillus oryzae Km-1-1 (pA2PEI) | (1) Cider; Wine | (1) Good Manufacturing Practice | ||
| (2) Single-strength fruit juices | (2) Good Manufacturing Practice | |||
| (3) Unstandardized fruit and vegetable products | (3) Good Manufacturing Practice | |||
| P.4 | Pentosanase | Aspergillus niger var.; Bacillus subtilis var. | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Corn for degermination | (2) Good Manufacturing Practice | |||
| (3) Distillers’ Mash | (3) Good Manufacturing Practice | |||
| (4) Mash destined for vinegar manufacture | (4) Good Manufacturing Practice | |||
| (5) Unstandardized bakery products | (5) Good Manufacturing Practice | |||
| (6) Bread; Flour; Whole wheat flour | (6) Good Manufacturing Practice | |||
| Trichoderma reesei (QM9414) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Distiller’s Mash | (2) Good Manufacturing Practice | |||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | |||
| P.5 | Pepsin | Glandular layer of porcine stomach | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Cheddar cheese; Cottage cheese; Cream cheese; Cream cheese spread; Cream cheese spread with (naming the added ingredients); Cream cheese with (naming the added ingredients); (naming the variety) Cheese | (2) Good Manufacturing Practice | |||
| (3) Defatted soya flour | (3) Good Manufacturing Practice | |||
| (4) Pre-cooked (instant) breakfast cereals | (4) Good Manufacturing Practice | |||
| (5) Hydrolyzed animal, milk and vegetable proteins | (5) Good Manufacturing Practice | |||
| P.5A | Phospholipase | Streptomyces violaceoruber | (1) Modified lecithin | (1) Good Manufacturing Practice |
| (2) Unstandardized egg products | (2) Good Manufacturing Practice | |||
| Aspergillus oryzae (pPFJo142) | Cheddar cheese; (naming the variety) Cheese | Good Manufacturing Practice | ||
| Aspergillus niger (PLA-54) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| (3) Unstandardized whole egg; unstandardized egg yolk | (3) Good Manufacturing Practice | |||
| (4) Modified lecithin | (4) Good Manufacturing Practice | |||
| P.6 | Protease | Aspergillus oryzae var.; Aspergillus niger var.; Bacillus subtilis var. | (1) Ale; Beer; Light beer; Malt liquor;Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bread; Flour; Whole wheat flour | (2) Good Manufacturing Practice | |||
| (3) Dairy based flavouring preparations | (3) Good Manufacturing Practice | |||
| (4) Distillers’ Mash | (4) Good Manufacturing Practice | |||
| (5) Sausage casings | (5) Good Manufacturing Practice | |||
| (6) Hydrolyzed animal, milk and vegetable protein | (6) Good Manufacturing Practice | |||
| (7) Industrial spray-dried cheese powder | (7) Good Manufacturing Practice | |||
| (8) Meat cuts | (8) Good Manufacturing Practice | |||
| (9) Meat tenderizing preparations | (9) Good Manufacturing Practice | |||
| (10) Pre-cooked (instant) breakfast cereals | (10) Good Manufacturing Practice | |||
| (11) Unstandardized bakery products | (11) Good Manufacturing Practice | |||
| (12) Cheddar cheese; Cheddar cheese for processing (granular curd cheese; Stirred curd cheese; Washed curd cheese); Colby cheese | (12) Good Manufacturing Practice | |||
| (13) Plant-based beverages | (13) Good Manufacturing Practice | |||
| Micrococcus caseolyticus var. | (1) (naming the variety) Cheese | (1) Good Manufacturing Practice | ||
| Bacillus licheniformis (Cx) | (1) Hydrolyzed animal, milk and vegetable protein | (1) Good Manufacturing Practice | ||
| P.7 | Pullulanase | Bacillus acidopullulyticus NCIB 11647; Bacillus licheniformis SE2-Pul-int211 (pUBCDEBR A11DNSI) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice |
| (2) Starch used in the production of dextrins, dextrose, glucose (glucose syrup), glucose solids (dried glucose syrup) or fructose syrups and solids, maltose | (2) Good Manufacturing Practice | |||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | |||
| Bacillus licheniformis BMP 139 (pR11Amp) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Brewers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Starch used in the production of dextrins, dextrose, glucose (glucose syrup), glucose solids (dried glucose syrup) or fructose syrups and solids, maltose | (3) Good Manufacturing Practice | |||
| (4) Unstandardized bakery products | (4) Good Manufacturing Practice | |||
| Bacillus subtilis B1-163 (pEB301) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Brewers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Distillers’ Mash | (3) Good Manufacturing Practice | |||
| (4) Starch used in the production of dextrins, dextrose, glucose (glucose syrup), glucose solids (dried glucose syrup) or fructose syrups and solids, maltose | (4) Good Manufacturing Practice | |||
| (5) Unstandardized bakery products | (5) Good Manufacturing Practice | |||
| Bacillus subtilis RB121 (pDG268) | (1) Brewers’ Mash | (1) Good Manufacturing Practice | ||
| (2) Distillers’ Mash | (2) Good Manufacturing Practice | |||
| (3) Starch used in the production of dextrins, dextrose, glucose (glucose syrup), glucose solids (dried glucose syrup) or maltose | (3) Good Manufacturing Practice | |||
| R.1 | Rennet | Aqueous extracts from the fourth stomach of calves, kids or lambs | (1) Cheddar cheese; Cottage cheese; Cream cheese; Cream cheese spread; Cream cheese spread with (naming the added ingredients); Cream cheese with (naming the added ingredients); (naming the variety) Cheese; Sour cream | (1) Good Manufacturing Practice |
| (2) Unstandardized milk based dessert preparations | (2) Good Manufacturing Practice | |||
| T.01 | Transglutaminase | Streptoverticillium mobaraense strain S-8112 | (1) Unstandardized prepared fish products | (1) Good Manufacturing Practice |
| (2) Simulated meat products | (2) Good Manufacturing Practice | |||
| (3) Unstandardized cheese products | (3) Good Manufacturing Practice | |||
| (4) Unstandardized processed cheese products | (4) Good Manufacturing Practice | |||
| (5) Unstandardized cream cheese products | (5) Good Manufacturing Practice | |||
| (6) Yogurt | (6) Good Manufacturing Practice | |||
| (7) Unstandardized frozen dairy desserts | (7) Good Manufacturing Practice | |||
| T.1 | Trypsin | Pancreas of the hog (Sus scrofa) | (1) Hydrolyzed animal, milk and vegetable proteins | (1) Good Manufacturing Practice |
| X.1 | Xylanase | Aspergillus oryzae Fa 1–1 (pA2X1TI) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| Aspergillus oryzae JaL 339 (pJaL537); Bacillus subtilis DIDK 0115 (pUB110 OIS2) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
| Bacillus subtilis CF 307 (pJHPaprE-xynAss-BS3xylanase#1) | (1) Bread; Flour; Whole wheat flour | (1) Good Manufacturing Practice | ||
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | |||
- SOR/78-402, s. 6
- SOR/78-876, s. 3
- SOR/79-662, ss. 14 to 17
- SOR/80-501, s. 4
- SOR/80-632, s. 5
- SOR/81-60, s. 11
- SOR/81-934, ss. 7 to 10
- SOR/82-383, s. 10
- SOR/82-566, s. 2
- SOR/82-1071, s. 17
- SOR/84-302, s. 4
- SOR/84-762, ss. 8, 9
- SOR/84-801, s. 4
- SOR/86-89, ss. 4 to 6
- SOR/86-1112, s. 4
- SOR/87-254, s. 2
- SOR/87-640, s. 7
- SOR/88-281, s. 1
- SOR/90-24, ss. 1 to 3
- SOR/90-87, ss. 10 to 12
- SOR/90-469, s. 3
- SOR/91-124, s. 5(F)
- SOR/91-487, s. 1
- SOR/91-691, s. 1
- SOR/92-63, s. 4
- SOR/92-94, s. 4
- SOR/92-195, s. 1
- SOR/92-197, s. 9
- SOR/92-231, s. 1
- SOR/92-518, s. 1
- SOR/92-591, s. 2(F)
- SOR/94-29, s. 1
- SOR/94-182, s. 1
- SOR/94-212, s. 9
- SOR/94-417, s. 2
- SOR/94-552, s. 1
- SOR/94-689, s. 2
- SOR/94-712, s. 1
- SOR/95-65, s. 1
- SOR/95-183, s. 9
- SOR/95-525, ss. 1, 2
- SOR/96-375, s. 1
- SOR/97-81, s. 1
- SOR/97-82, s. 1
- SOR/97-122, ss. 4(F), 5
- SOR/97-508, ss. 1, 2
- SOR/97-513, s. 1
- SOR/97-558, s. 4
- SOR/98-454, s. 1
- SOR/98-458, ss. 6, 7(F)
- SOR/2000-336, ss. 3 to 5
- SOR/2000-417, s. 4
- SOR/2003-130, s. 4
- SOR/2004-84, s. 1
- SOR/2005-98, ss. 4 to 7, 8(F)
- SOR/2005-394, ss. 1 to 6
- SOR/2007-225, s. 1
- SOR/2010-41, ss. 1 to 6, 9(E)
- SOR/2010-42, ss. 1 to 4
- SOR/2010-94, s. 8(E)
- SOR/2010-142, s. 17
- SOR/2010-143, ss. 14 to 26
- SOR/2012-26, s. 4
- SOR/2012-45, s. 1
- SOR/2012-46, s. 4
- SOR/2012-104, ss. 10, 21
TABLE VI
Food Additives That May Be Used as Firming Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Aluminum Sulphate | (1) Canned crabmeat, lobster, salmon, shrimp and tuna; Pickles and relishes | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.2 | Ammonium Aluminum Sulphate | (1) Pickles and relishes | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.1 | Calcium Chloride | (1) Canned apples | (1) 0.026% calculated as calcium |
| (2) Canned grapefruit | (2) 0.035% of the total calcium content, whether added or otherwise present | ||
| (3) (naming the variety) cheese; Cheddar cheese | (3) 0.02% of the milk and milk products used | ||
| (4) Cottage cheese | (4) Good Manufacturing Practice | ||
| (5) Glaze of frozen fish | (5) Good Manufacturing Practice | ||
| (6) Olives | (6) 1.5% of the brine | ||
| (7) Pickles and relishes | (7) 0.4% | ||
| (8) Tomatoes; Canned vegetables (naming the vegetable); Frozen apples | (8) 0.026% calculated as calcium, and in the case of canned peas 0.035% calculated as calcium | ||
| (9) Unstandardized foods | (9) Good Manufacturing Practice | ||
| (10) Canned apricots | (10) 0.022% calculated as calcium | ||
| C.2 | Calcium Citrate | (1) Tomatoes; Canned vegetables; Frozen apples; Frozen sliced apples | (1) 0.026% calculated as calcium |
| (2) Canned apples | (2) 0.026% calculated as calcium | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| C.3 | Calcium Gluconate | Unstandardized foods | Good Manufacturing Practice |
| C.3A | Calcium Lactate | (1) Canned grapefruit | (1) 0.035% of the total calcium content, whether added or otherwise present |
| (2) Canned peas | (2) 0.035% calculated as calcium | ||
| C.4 | Calcium Phosphate, dibasic | Unstandardized foods | Good Manufacturing Practice |
| C.5 | Calcium Phosphate, monobasic | (1) Tomatoes; Canned vegetables; Frozen apples | (1) 0.026% calculated as calcium |
| (2) Canned apples | (2) 0.026% calculated as calcium | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| C.6 | Calcium Sulphate | (1) Tomatoes; Canned vegetables; Frozen apples | (1) 0.026% calculated as calcium |
| (2) Canned apples | (2) 0.026% calculated as calcium | ||
| P.1 | Potassium Aluminum Sulphate | (1) Pickles and relishes | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.1 | Sodium Aluminum Sulphate | (1) Pickles and relishes | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice |
- SOR/78-402, s. 7
- SOR/79-660, ss. 11, 12
- SOR/79-752, s. 7
- SOR/93-445, s. 1
- SOR/94-689, s. 2(F)
- SOR/2007-302, s. 4(F)
- SOR/2010-94, s. 8(E)
- SOR/2012-43, ss. 29, 30
TABLE VII
Food Additives That May Be Used as Glazing and Polishing Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acetylated Monoglycerides | (1) Unstandardized confectionery | (1) 0.4% |
| (2) Frozen fish | (2) Good Manufacturing Practice | ||
| B.1 | Beeswax | Unstandardized confectionery | 0.4% |
| C.1 | Carnauba Wax | Unstandardized confectionery | 0.4% |
| C.2 | Candelilla Wax | Unstandardized confectionery | 0.4% |
| G.1 | Gum Arabic | Unstandardized confectionery | 0.4% |
| G.2 | Gum Benzoin | Unstandardized confectionery | 0.4% |
| M.1 | Magnesium Silicate | Unstandardized confectionery | 0.4% |
| M.2 | Mineral Oil | Unstandardized confectionery | 0.15% |
| P.1 | Petrolatum | Unstandardized confectionery | 0.15% |
| S.1 | Shellac | Cake decorations; Unstandardized confectionery | 0.4% |
| S.2 | Spermaceti Wax | Unstandardized confectionery | 0.4% |
| Z.1 | Zein | Unstandardized confectionery | 1.0% |
- SOR/94-689, s. 2(F)
- SOR/2010-142, ss. 18 to 29
TABLE VIII
Miscellaneous Food Additives
| Item No. | Column I | Column II | Column III | Column IV |
|---|---|---|---|---|
| Additive | Permitted in or Upon | Purpose of Use | Maximum Level of Use | |
| A.01 | Acacia Gum | Ale; Beer; Light beer; Malt liquor; Porter; Stout; Wine | Fining agent | Good Manufacturing Practice |
| A.1 | Acetylated Monoglycerides | Unstandardized foods | Coating; Release agent | Good Manufacturing Practice |
| A.1.01 | Agar | Wine | Fining agent | Good Manufacturing Practice |
| A.1.1 | Aluminum Sulphate | Dried egg-white (dried albumen); Dried whole egg; Dried yolk; Frozen egg-white (frozen albumen); Frozen whole egg; Frozen yolk; Liquid egg-white (liquid albumen); Liquid whole egg; Liquid yolk | To stabilize albumen during pasteurization | 0.036% |
| A.2 | Ammonium Persulphate | Brewer’s yeast | Antimicrobial agent | 0.1% |
| A.3 | [Repealed, SOR/93-276, s. 4] | |||
| A.4 | [Repealed, SOR/93-276, s. 5] | |||
| B.2 | Beeswax | Unstandardized foods | Antisticking agent | 0.4% |
| B.2.1 | Benzoyl Peroxide | Liquid whey destined for the manufacture of dried whey products other than those for use in infant formula | To decolourize | 100 p.p.m. |
| B.3 | Brominated vegetable oil | (Naming the flavour) Flavour for use in beverages containing citrus or spruce oils | Density adjusting agent | 15 p.p.m. in beverages containing citrus or spruce oils as consumed |
| B.4 | n-Butane | Edible vegetable oil-based or lecithin-based pan coatings or a mixture of both | Propellant | Good Manufacturing Practice |
| C.1 | Caffeine | Cola type beverages | To characterize the product | 200 p.p.m. in the finished product |
| C.2 | Caffeine Citrate | Cola type beverages | To characterize the product | 200 p.p.m. calculated as caffeine, in the finished product |
| C.3 | Calcium Carbonate | (1) Flour; Whole wheat flour | (1) Carrier of benzoyl peroxide | (1) 900 p.p.m., in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| (2) [Repealed, SOR/94-227, s. 5] | ||||
| (3) Unstandardized confectionery | (3) Creaming and fixing agent | (3) Good Manufacturing Practice | ||
| (4) Chewing gum | (4) Filler | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Carrier and dusting agent | (5) Good Manufacturing Practice | ||
| C.3.01 | Calcium Chloride | Sausage having an edible coating | To stabilize the edible coating | Good Manufacturing Practice |
| C.3A | Calcium Lactate | (1) Egg albumen (delysozymized) | (1) Restoration of functional properties | (1) Good Manufacturing Practice (Quantity of calcium added not to exceed that lost during processing) |
| (2) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (2) To modify texture | (2) Good Manufacturing Practice | ||
| (3) Sausage having an edible coating | (3) To stabilize the edible coating | (3) Good Manufacturing Practice | ||
| C.4 | Calcium Oxide | (1) Frozen crustaceans and molluscs | (1) To facilitate the removal of extraneous matter and to reduce moisture loss during cooking | (1) When used in combination with sodium chloride (salt) and sodium hydroxide in solution, calcium oxide not to exceed 30 p.p.m. |
| (2) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (2) To modify texture | (2) Good Manufacturing Practice | ||
| C.5 | Calcium Phosphate dibasic | (1) Flour; Whole wheat flour | (1) Carrier of benzoyl peroxide | (1) 900 p.p.m. in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| (2) [Repealed, SOR/94-227, s. 6] | ||||
| C.6 | Calcium Phosphate, tribasic | (1) Flour; Whole wheat flour | (1) Carrier of benzoyl peroxide | (1) 900 p.p.m. in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| (2) [Repealed, SOR/94-227, s. 7] | ||||
| (3) Liquid whey destined for the manufacture of dried whey products other than those for use in infant formula | (3) Carrier of benzoyl peroxide | (3) 0.04% of dried whey product | ||
| (4) Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredient); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients) | (4) To improve colour, texture, consistency and spreadability | (4) 1.0% | ||
| C.7 | Calcium Silicate | Oil-soluble annatto | Carrier | Good Manufacturing Practice |
| C.8 | Calcium Stearate | Unstandardized confectionery | Release agent | Good Manufacturing Practice |
| C.9 | Calcium Stearoyl- 2-Lactylate | (1) Frozen egg-white (frozen albumen); Liquid egg-white (liquid albumen) | (1) Whipping agent | (1) 0.05% |
| (2) Dried egg-white (dried albumen) | (2) Whipping agent | (2) 0.5% | ||
| (3) Vegetable fat toppings | (3) Whipping agent | (3) 0.3% | ||
| (4) Dehydrated potatoes | (4) Conditioning agent | (4) 0.2% of dry weight | ||
| C.10 | Calcium Sulphate | (1) Flour, Whole wheat flour | (1) Carrier of benzoyl peroxide | (1) 900 p.p.m. in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| (2) [Repealed, SOR/94-227, s. 8] | ||||
| (3) Baking powder | (3) Neutral filler | (3) Good Manufacturing Practice | ||
| (4) Liquid whey destined for the manufacture of dried whey products, other than those for use in infant formula | (4) Carrier of benzoyl peroxide | (4) 0.3% of the dried whey product | ||
| C.11 | Carbon Dioxide | (1) Ale; Beer; Carbonated (naming the fruit) juice; Cider; Light beer; Malt liquor; Porter; Stout; Wines; Water represented as mineral water or spring water | (1) Carbonation | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Carbonation and pressure dispensing agent | (2) Good Manufacturing Practice | ||
| (3) Cottage Cheese; Creamed Cottage Cheese | (3) To extend durable life | (3) Good Manufacturing Practice | ||
| C.11.1 | Carboxymethyl Cellulose, cross-linked (Sodium Carboxymethyl Cellulose, cross-linked) | Table-top sweetener tablets that contain acesulfame-potassium, aspartame, erythritol, neotame or sucralose | Tablet disintegration | Good Manufacturing Practice |
| C.12 | Castor Oil | Unstandardized confectionery | Release agent | Good Manufacturing Practice |
| C.13 | [Repealed, SOR/2010-142, s. 35] | |||
| C.13.1 | Cellulose, Powdered | (1) Batter and breading | (1) Bulking agent | (1) 1% |
| (2) Canapé toast | (2) Bulking agent | (2) 2% | ||
| (3) Unstandardized confectionery that meet the conditions set out in column 2 of item 3 of the table following section B.01.513 for the subject “Reduced in energy” set out in column 1 | (3) Bulking agent | (3) 25% | ||
| (4) Unstandardized edible ices | (4) Bulking agent | (4) 3% | ||
| (5) Fillings | (5) Bulking agent | (5) 0.5% | ||
| (6) Foods sold in tablet form | (6) Bulking agent | (6) 50% | ||
| (7) Icings | (7) Bulking agent | (7) 1% | ||
| (8) Seasonings | (8) Bulking agent | (8) 3% | ||
| (9) Sweet baked goods | (9) Bulking agent | (9) 8% | ||
| C.14A | Chloropentafluo-roethane | Unstandardized foods | Pressure dispensing and aerating agent | Good Manufacturing Practice |
| C.15 | Citric Acid | (1) Beef blood | (1) Anticoagulant | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Culture nutrient | (2) Good Manufacturing Practice | ||
| C.16 | Copper gluconate | Breath freshener products | To characterize the product | 50 p.p.m. |
| C.17 | Copper Sulphate | Wine | Fining agent | 0.0001%, calculated as copper, in the finished product |
| D.1 | Dimethylpolysiloxane Formulations | (1) Apple (or rhubarb) and (naming the fruit) Jam; Fats and oils; Fig marmalade; Fig marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Pineapple marmalade; Pineapple marmalade with pectin; Reconstituted lemon juice; Reconstituted lime juice; Shortening; Skim milk powder; Wine | (1) Antifoaming agent | (1) 10 p.p.m. of dimethylpolysiloxane |
| (2) Pineapple juice; Blends of pineapple juice with other fruit juices; canned pineapple (when pineapple juice is used as the packing medium) | (2) Anti-foaming agent | (2) 10 p.p.m. of dimethylpolysiloxane | ||
| (3) Surfaces that come in contact with food | (3) Release agent | (3) Good Manufacturing Practice (Residue of dimethylpolysiloxane in food not to exceed 10 p.p.m.) | ||
| (4) Unstandardized foods | (4) Antifoaming agent | (4) 10 p.p.m. of dimethylpolysiloxane | ||
| (5) Wort used in the manufacture of Ale, Beer, Light beer, Malt liquor, Porter and Stout | (5) Antifoaming agent | (5) 10 p.p.m. of dimethylpolysiloxane | ||
| D.3 | Dioctyl sodium sulfosuccinate | (1) Fumaric acid-acidulated dry beverage bases | (1) Wetting agent | (1) 10 p.p.m. in the finished drink |
| (2) Sausage casings | (2) Reduce casing breakage | (2) 200 p.p.m. of the casing | ||
| E.1 | Ethoxyquin | Paprika; Ground chili pepper | To promote colour retention | 100 p.p.m. |
| E.2 | [Repealed, SOR/2016-143, s. 1] | |||
| F.1 | Ferrous Gluconate | Ripe olives | Colour retention | Good Manufacturing Practice |
| G.1 | Gelatin | Beer; Cider; Wine | Fining agent | Good Manufacturing Practice |
| G.2 | [Repealed, SOR/89-175, s. 2] | |||
| G.2A | Glucono delta lactone | (1) Cooked sausage, Meat Loaf | (1) To accelerate colour fixing | (1) 0.5% |
| (2) Dry Sausage | (2) To assist in curing | (2) Good Manufacturing Practice | ||
| G.3 | Glycerol | (1) Meat curing compounds; Sausage casings | (1) Humectant | (1) Good Manufacturing Practice |
| (2) Preserved meats (Division 14) | (2) Glaze for preserved meats | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Humectant; Plasticizer | (3) Good Manufacturing Practice | ||
| G.4 | Glycerol ester of wood rosin | Beverages containing citrus or spruce oils | Density adjusting agent | 100 p.p.m. |
| H.1 | Hydrogen Peroxide | (1) Brewers’ mash | (1) Clarification aid | (1) 135 p.p.m. in the mash |
| (2) Liquid whey destined for the manufacture of dried whey products | (2) To decolourize and maintain pH | (2) 100 p.p.m. (see also subitem C.1(3) of Table V) | ||
| (3) Oat hulls used in the manufacture of oat hull fibre | (3) Bleaching agent | (3) Good Manufacturing Practice | ||
| I | Isobutane | Edible vegetable oil-based or lecithin-based pan coatings or a mixture of both | Propellant | Good Manufacturing Practice |
| L.1 | Lactylic Esters of Fatty Acids | Unstandardized foods | Plasticizing agent | Good Manufacturing Practice |
| L.2 | Lanolin | Chewing gum | Plasticizing agent | Good Manufacturing Practice |
| L.3 | Lecithin | (1) Surfaces that come in contact with food | (1) Release agent | (1) Good Manufacturing Practice |
| (2) Infant cereal products | (2) Release agent | (2) 1.75% as consumed | ||
| L.4 | L-Leucine | Table-top sweetener tablets that contain acesulfame-potassium, aspartame, erythritol, neotame or sucralose | Lubricant or binder in tablet manufacture | Good Manufacturing Practice |
| M.1 | Magnesium Aluminum Silicate | Chewing gum | Dusting agent | Good Manufacturing Practice |
| M.2 | Magnesium Carbonate | (1) Flour, Whole wheat flour | (1) Carrier of benzoyl peroxide | (1) 900 p.p.m. in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| (2) [Repealed, SOR/94-227, s. 9] | ||||
| (3) Unstandardized confectionery | (3) Release agent | (3) Good Manufacturing Practice | ||
| M.2A | Magnesium Chloride | Egg albumen (delysozymized) | Restoration of functional properties | Good Manufacturing Practice (Quantity of magnesium added not to exceed that lost during processing) |
| M.3 | Magnesium Silicate | (1) Unstandardized confectionery | (1) Release agent | (1) Good Manufacturing Practice |
| (2) Chewing gum | (2) Dusting agent | (2) Good Manufacturing Practice | ||
| (3) Rice | (3) Coating | (3) Good Manufacturing Practice | ||
| M.4 | Magnesium Stearate | (1) Unstandardized confectionery | (1) Release agent | (1) Good Manufacturing Practice |
| (2) Foods sold in tablet form | (2) Binding agent | (2) Good Manufacturing Practice | ||
| M.4A | Magnesium Sulphate | Egg albumen (delysozymized) | Restoration of functional properties | Good Manufacturing Practice (Quantity of magnesium added not to exceed that lost during processing) |
| M.5A | [Repealed, SOR/93-276, s. 6] | |||
| M.5C | Methyl Ethyl Cellulose | Unstandardized foods | Aerating agent | Good Manufacturing Practice |
| M.6 | Microcrystalline Cellulose | (1) Ice milk mix | (1) Bodying and texturizing agent | (1) 1.5% |
| (2) Sherbet | (2) Bodying and texturizing agent | (2) 0.5% | ||
| (3) Unstandardized foods that meet the conditions set out in column 2 of item 3 of the table following section B.01.513 for the subject “Reduced in energy” set out in column 1 | (3) Filler | (3) Good Manufacturing Practice | ||
| (4) Whipped vegetable oil topping | (4) Bodying and texturizing agent | (4) 1.5% | ||
| (5) Unstandardized frozen desserts | (5) Bodying and texturizing agent | (5) 0.5% | ||
| (6) Unstandardized sandwich spreads; Unstandardized dips | (6) Bodying and texturizing agent | (6) 3.0% | ||
| (7) Unstandardized foods other than those unstandardized foods referred to in this item | (7) Bodying and texturizing agent | (7) 2.0% | ||
| (8) Ice cream mix | (8) Bodying and texturizing agent | (8) 0.5% or, if used in combination with stabilizing agents, the combined amount shall not exceed 0.5% of the ice cream made from the mix | ||
| (9) Table-top sweetener tablets containing aspartame | (9) Tablet disintegration | (9) 2.2% | ||
| (10) Cream for whipping | (10) Stabilizing and thickening agent | (10) 0.2% | ||
| (11) Breath freshener products | (11) Bodying and texturizing agent | (11) 9.0% | ||
| M.7 | Mineral Oil | (1) Bakery products; Seeded raisins; Unstandardized confectionery | (1) Release agent | (1) 0.3% in accordance with section B.01.047. If petrolatum is also used as a release agent in bakery products the total of any combination of petrolatum and mineral oil must not exceed 0.15% |
| (2) Fresh fruits and vegetables | (2) Coating | (2) 0.3% in accordance with section B.01.047 | ||
| (3) Sausage casings | (3) Lubricant | (3) 5% in accordance with paragraph B.01.047(e) (Residues of mineral oil in a raw sausage prepared with such casings not to exceed 200 p.p.m.; in cooked sausage, 30 p.p.m.) | ||
| (4) Salt Substitute | (4) Binding agent and protective coating | (4) 0.6% in accordance with paragraph B.01.047(h) | ||
| M.8 | Monoacetin | Unstandardized bakery products | Plasticizer | Good Manufacturing Practice |
| M.9 | Mono- and di-glycerides | (1) Apple (or rhubarb) and (naming the fruit) Jam; Fats and oils; Fig marmalade; Fig marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Pineapple marmalade; Pineapple marmalade with pectin | (1) Antifoaming agent | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Antifoaming agent; Humectant; Release agent | (2) Good Manufacturing Practice | ||
| M.10 | Mono-glycerides | Unstandardized foods | Antifoaming agent; Humectant; Release agent | Good Manufacturing Practice |
| N.1 | Nitrogen | (1) Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients) | (1) To improve spreadability | (1) Good Manufacturing Practice |
| (2) Margarine | (2) To improve spreadability | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Pressure dispensing agent | (3) Good Manufacturing Practice | ||
| N.2 | Nitrous Oxide | Unstandardized foods | Pressure dispensing agent | Good Manufacturing Practice |
| O.1 | Octafluorocyclobu-tane | Unstandardized foods | Pressure dispensing and aerating agent | Good Manufacturing Practice |
| O.2 | Oxystearin | Cotton seed oil; Peanut oil; Soy bean oil | To inhibit crystal formation | 0.125% |
| O.3 | Ozone | (1) Cider | (1) Maturing agent | (1) Good Manufacturing Practice |
| (2) Water represented as mineral water or spring water | (2) Chemosterilant | (2) Good Manufacturing Practice | ||
| (3) Wine | (3) Maturing agent | (3) Good Manufacturing Practice | ||
| P.1 | Pancreas Extract | Acid producing bacterial cultures | To control bacteriophages | Good Manufacturing Practice |
| P.1A | Paraffin Wax | (1) Fresh fruits and vegetables | (1) Coating | (1) 0.3% in accordance with section B.01.047 |
| (2) Cheese and turnips | (2) Coating | (2) Good Manufacturing Practice in accordance with section B.01.047 | ||
| P.2 | Petrolatum | (1) Bakery products | (1) Release agent | (1) 0.15% in accordance with section B.01.047. If mineral oil is also used as a release agent the total of any combination of petrolatum and mineral oil must not exceed 0.15% |
| (2) Fresh fruits and vegetables | (2) Coating | (2) 0.3% in accordance with section B.01.047 | ||
| P.2A | Polyethylene glycol (molecular weight 3000-9000) | (1) Soft drinks | (1) Antifoaming agent | (1) 10 p.p.m. |
| (2) Table-top sweetener tablets containing aspartame | (2) Lubricant | (2) 1.0% | ||
| (3) L-Lysine tablets | (3) Tablet binder | (3) 7.0% | ||
| P.2B | Polydextrose | Unstandardized foods | Bodying and texturizing agent | Good Manufacturing Practice |
| P.3 | Polyvinylpyrroli-done | (1) Ale; Beer; Cider; Light beer; Malt liquor; Porter; Stout; Wine | (1) Fining agent | (1) 2 p.p.m. in the finished product |
| (2) Table-top sweetener tablets containing aspartame | (2) Tablet binder | (2) 0.3% | ||
| (3) Colour lake dispersions for use in unstandardized confectionery in tablet form | (3) Viscosity reduction agent and stabilizer in colour lake dispersions | (3) Good Manufacturing Practice (Residues of polyvinyl pyroliodone not to exceed 100 p.p.m. in the finished foods) | ||
| P.4 | Potassium Aluminum Sulphate | Flour; Whole wheat flour | Carrier of benzoyl peroxide | 900 p.p.m., in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| P.4.1 | Potassium Ferrocyanide | Wine | Fining agent | Good Manufacturing Practice |
| P.5 | Potassium Stearate | (1) Chewing gum | (1) Plasticizing agent | (1) Good Manufacturing Practice |
| (2) Emulsifying preparations containing propylene glycol monoesters | (2) Stabilizing agent | (2) 2% | ||
| P.6 | Propane | Unstandardized foods | Pressure dispensing and aerating agent | Good Manufacturing Practice |
| P.7 | Propylene Glycol | (1) Oil-soluble annatto | (1) Solvent | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Humectant | (2) Good Manufacturing Practice | ||
| Q.1 | Quillaia Extract | Beverage bases; Beverage mixes; Soft drinks | Foaming Agent | Good Manufacturing Practice |
| S.1 | Saponin | Beverage bases; Beverage mixes; Soft drinks | Foaming agent | Good Manufacturing Practice |
| S.1.01 | Silicon Dioxide | Edible vegetable oil-based cookware coating emulsions | Suspending agent | 2.0% of preparation |
| S.1.1 | Sodium Acid Pyrophosphate | Frozen fish fillets; frozen minced fish; frozen lobster; frozen crab; frozen clams; frozen shrimp | To reduce processing losses and to reduce thaw drip | Used in combination with sodium tripolyphosphate and sodium pyrophosphate tetrabasic, total added pyrophosphate not to exceed 0.5% calculated as sodium phosphate, dibasic |
| S.2 | Sodium Aluminum Sulphate | Flour; Whole wheat flour | Carrier of benzoyl peroxide | 900 p.p.m. in accordance with subparagraphs B.13.001(e)(vi) and B.13.005(d)(vi) |
| S.3 | Sodium Bicarbonate | (1) Unstandardized confectionery | (1) Aerating agent | (1) Good Manufacturing Practice |
| (2) Salt | (2) To stabilize potassium iodide in salt | (2) Good Manufacturing Practice | ||
| S.3A | Sodium Carbonate | In combination with sodium hexametophosphate for use on frozen fish fillets, frozen lobster, frozen crabs, frozen clams and frozen shrimp | To reduce thaw drip | 15% of the combination of sodium carbonate and sodium hexametaphosphate |
| S.3B | Sodium Carboxymethyl Cellulose | Sausage casings | Coating to enable peeling | 0.25% of the casing |
| S.4 | Sodium Citrate | (1) Beef blood | (1) Anticoagulant | (1) 0.5% |
| (2) Sour cream | (2) Flavour precursor | (2) 0.1% | ||
| (3) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (3) To modify texture | (3) Good Manufacturing Practice | ||
| S.5 | Sodium Ferrocyanide Decahydrate | Dendritic salt | As an adjuvant in the production of dendritic salt crystals | 13 p.p.m. calculated as anhydrous sodium ferrocyanide |
| S.6 | Sodium Hexametaphosphate | (1) Beef blood | (1) Anti-coagulant | (1) 0.2% |
| (2) Frozen fish fillets; frozen lobsters; frozen crab; frozen clams and frozen shrimp | (2) To reduce thaw drip | (2) 0.5% total added phosphate calculated as sodium phosphate, dibasic | ||
| (3) Gelatin intended for marshmallow compositions | (3) Whipping agent | (3) 2% | ||
| S.6A | Sodium Hydroxide | Frozen crustaceans and molluscs | To facilitate the removal of extraneous matter and to reduce moisture loss during cooking | When used in combination with sodium chloride (salt) and calcium oxide in solution, sodium hydroxide not to exceed 70 p.p.m. |
| S.6.1 | Sodium Lauryl Sulphate | (1) Dried egg-white (dried albumen) | (1) Whipping agent | (1) 0.1% |
| (2) Frozen egg-white (frozen albumen); Liquid egg-white (liquid albumen) | (2) Whipping agent | (2) 0.0125% | ||
| (3) Gelatin intended for marshmallow compositions | (3) Whipping agent | (3) 0.5% | ||
| S.6.2 | Sodium Potassium Copper Chlorophyllin | Breath freshener products | To characterize the product | 700 p.p.m. |
| S.7 | Sodium Phosphate, dibasic | (1) Frozen fish | (1) To prevent cracking of glaze | (1) Good Manufacturing Practice |
| (2) Frozen mushrooms | (2) To prevent discolouration | (2) Good Manufacturing Practice | ||
| S.7.1 | Sodium Pyrophosphate, tetrabasic | Frozen fish fillets; frozen minced fish; frozen lobster; frozen crab; frozen clams; frozen shrimp | To reduce processing losses and to reduce thaw drip | Used in combination with sodium tripolyphosphate and sodium acid pyrophosphate, total added phosphate not to exceed 0.5% calculated as sodium phosphate, dibasic |
| S.8 | Sodium Silicate | Canned drinking water | Corrosion inhibitor | Good Manufacturing Practice |
| S.9 | Sodium Stearate | Chewing gum | Plasticizing agent | Good Manufacturing Practice |
| S.9A | Sodium Stearoyl- 2-Lactylate | (1) Frozen egg-white (frozen albumen); Liquid egg-white (liquid albumen) | (1) Whipping agent | (1) 0.05% |
| (2) Dried egg-white (dried albumen) | (2) Whipping agent | (2) 0.5% | ||
| (3) Oil toppings or topping mixes | (3) Whipping agent | (3) 0.3% | ||
| (4) Dehydrated potatoes | (4) Conditioning agent | (4) 0.2% of dry weight | ||
| S.9B | Sodium Sulphate | Frozen mushrooms | To prevent discolouration | Good Manufacturing Practice |
| S.9C | Sodium Sulphite | Canned flaked tuna | To prevent discolouration | 300 p.p.m. |
| S.10 | Sodium Thiosulphate | Salt | To stabilize potassium iodine in salt | Good Manufacturing Practice |
| S.11 | Sodium Tripolyphosphate | Frozen fish fillets; frozen minced fish; frozen comminuted fish; frozen lobster; frozen crab; frozen clams and frozen shrimp | To reduce processing losses and to reduce thaw drip | Used singly or in combination with sodium acid pyrophosphate and sodium pyrophosphate tetrabasic, total added phosphate not to exceed 0.5% calculated as sodium phosphate, dibasic |
| S.12 | [Repealed, SOR/93-276, s. 8] | |||
| S.13 | Stannous Chloride | (1) Asparagus packed in glass containers or fully lined (lacquered) cans | (1) Flavour and colour stabilizer | (1) 25 p.p.m. calculated as tin |
| (2) Canned carbonated soft drinks; concentrated fruit juices except frozen concentrated orange juice; lemon juice; lime juice | (2) Flavour and colour stabilizer | (2) Good Manufacturing Practice | ||
| S.14 | Stearic Acid | (1) Unstandardized confectionery | (1) Release agent | (1) Good Manufacturing Practice |
| (2) Chewing gum | (2) Plasticizing agent | (2) Good Manufacturing Practice | ||
| (3) Foods sold in tablet form | (3) Release agent and lubricant | (3) Good Manufacturing Practice | ||
| S.15 | Sodium Methyl Sulphate | Pectin | A processing aid, the result of methylation of pectin by sulfuric acid and methyl alcohol and neutralized by sodium bicarbonate | 0.1% of pectin |
| S.15A | [Repealed, SOR/93-276, s. 9] | |||
| S.16 | Sucrose Acetate Isobutyrate | (Naming the flavour) Flavour for use in beverages containing citrus or spruce oils | Density adjusting agent | 300 p.p.m. in beverages containing citrus or spruce oils as consumed |
| S.17 | Sulphuric Acid | Coffee beans | To improve the extraction yield of coffee solids | Good Manufacturing Practice |
| T.1 | Talc | (1) Dried split legumes; Rice | (1) Coating agent | (1) Good Manufacturing Practice |
| (2) Chewing gum base | (2) Filler | (2) Good Manufacturing Practice | ||
| (3) Chewing gum | (3) Dusting agent | (3) Good Manufacturing Practice | ||
| T.2 | Tannic Acid | (1) Chewing gum | (1) To reduce adhesion | (1) Good Manufacturing Practice |
| (2) Cider; Honey wine; Wine | (2) Fining agent | (2) 200 p.p.m | ||
| T.2A | [Repealed, SOR/93-276, s. 10] | |||
| T.3 | Triacetin | Cake mixes | Wetting agent | Good Manufacturing Practice |
| T.4 | Triethyl Citrate | Dried egg-white (dried albumen); Frozen egg-white (frozen albumen); Liquid egg-white (liquid albumen) | Whipping agent | 0.25% |
| X.1 | [Repealed, SOR/93-276, s. 11] | |||
- SOR/78-401, s. 3
- SOR/78-403, ss. 23 to 25
- SOR/78-876, s. 4
- SOR/79-660, s. 13
- SOR/79-752, s. 8
- SOR/80-632, ss. 6 to 13
- SOR/81-83, s. 4
- SOR/81-617, s. 3
- SOR/81-934, ss. 11, 12
- SOR/82-566, ss. 3, 4
- SOR/82-1071, ss. 18 to 20
- SOR/83-410, s. 3
- SOR/83-584, s. 1
- SOR/83-932, ss. 5, 6
- SOR/84-17, s. 6
- SOR/84-441, s. 1
- SOR/84-602, s. 3
- SOR/84-762, s. 10
- SOR/84-801, ss. 5, 6
- SOR/86-1112, s. 5
- SOR/86-1125, s. 2
- SOR/87-469, s. 1
- SOR/87-640, s. 8
- SOR/88-419, s. 4
- SOR/88-534, ss. 5, 6
- SOR/89-175, s. 2
- SOR/89-197, s. 1
- SOR/89-198, s. 11
- SOR/89-555, ss. 2, 3
- SOR/91-90, s. 2
- SOR/91-124, ss. 6 to 9
- SOR/91-149, s. 3
- SOR/91-186, s. 1
- SOR/91-409, s. 7
- SOR/91-527, s. 4
- SOR/92-229, s. 1
- SOR/92-344, ss. 2 to 4
- SOR/93-276, ss. 4 to 11
- SOR/94-416, s. 1
- SOR/94-227, ss. 5 to 10
- SOR/94-689, s. 2(F)
- SOR/94-723, s. 1
- SOR/96-260, s. 1
- SOR/96-378, s. 1
- SOR/97-509, s. 1
- SOR/98-580, s. 1(F)
- SOR/99-97, s. 1
- SOR/99-420, s. 11(F)
- SOR/2000-353, s. 8(F)
- SOR/2001-94, s. 3
- SOR/2005-316, ss. 2(F), 3
- SOR/2006-91, ss. 6 to 12
- SOR/2007-75, s. 7
- SOR/2010-41, s. 9(E)
- SOR/2010-94, s. 8(E)
- SOR/2010-142, ss. 30(F), 31 to 39, 40(F), 41 to 45, 46(F), 47 to 51, 59(F)
- SOR/2010-143, ss. 27 to 31
- SOR/2011-235, s. 16(F)
- SOR/2011-279, s. 1
- SOR/2011-282, s. 2
- SOR/2012-43, s. 31(F)
- SOR/2012-44, ss. 1, 2
- SOR/2012-105, s. 1
- SOR/2012-111, ss. 2, 3
- SOR/2016-143, s. 1
TABLE IX
Food Additives That May Be Used as Sweeteners
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or on | Maximum Level of Use | |
| A.01 | Acesulfame-potassium | (1) Table-top sweeteners | (1) Good Manufacturing Practice |
| (2) Carbonated beverages | (2) 0.025% in beverages as consumed | ||
| (3) Unstandardized beverage concentrates; Unstandardized beverage mixes; Unstandardized beverages; Unstandardized dairy beverages | (3) 0.05% in beverages as consumed | ||
| (4) Filling mixes; Fillings; Topping mixes; Toppings; Unstandardized dessert mixes; Unstandardized desserts; Yogurt | (4) 0.1% in products as consumed | ||
| (5) Chewing gum; Breath freshener products | (5) 0.35% | ||
| (6) Unstandardized fruit spreads | (6) 0.1% | ||
| (7) Unstandardized salad dressings | (7) 0.05% | ||
| (8) Unstandardized confectionery | (8) 0.25% | ||
| (9) Baking mixes; Unstandardized bakery products | (9) 0.1% in products as consumed | ||
| (10) Canned (naming the fruit); Unstandardized canned fruit | (10) 0.007% | ||
| A.1 | Aspartame | (1) Table-top sweeteners | (1) Good Manufacturing Practice |
| (2) Breakfast cereals | (2) 0.5% | ||
| (3) Unstandardized beverage concentrates; Unstandardized beverage mixes; Unstandardized beverages | (3) 0.1% in beverages as consumed | ||
| (4) Filling mixes; Fillings; Topping mixes; Toppings; Unstandardized dessert mixes; Unstandardized desserts; Yogurt | (4) 0.3% in products as consumed | ||
| (5) Chewing gum; Breath freshener products | (5) 1.0% | ||
| (6) Unstandardized fruit spreads; Unstandardized purées; Unstandardized sauces; Unstandardized table syrups | (6) 0.2% | ||
| (7) Nut spreads; Peanut spreads; Unstandardized salad dressings | (7) 0.05% | ||
| (8) Unstandardized condiments | (8) 0.2% | ||
| (9) Confectionery glazes for snack foods; Sweetened seasonings or coating mixes for snack foods | (9) 0.1% | ||
| (10) Unstandardized confectionery; Unstandardized confectionery coatings | (10) 0.3% | ||
| A.2 | Aspartame, encapsulated to prevent degradation during baking | Baking mixes; Unstandardized bakery products | 0.4% in product as consumed |
| E.1 | Erythritol | (1) Table-top sweeteners | (1) Good Manufacturing Practice |
| (2) Dietetic beverages | (2) 3.5% | ||
| (3) Fat-based cream fillings and toppings | (3) 60% | ||
| (4) Dietetic cookies and wafers | (4) 7% | ||
| (5) Soft candies | (5) 40% | ||
| (6) Hard candies | (6) 50% | ||
| (7) Chewing gum | (7) 60% | ||
| H.1 | Hydrogenated starch hydrolysates | Unstandardized foods | Good Manufacturing Practice |
| I.1 | Isomalt | Unstandardized foods | Good Manufacturing Practice |
| L.1 | Lactitol | Unstandardized foods | Good Manufacturing Practice |
| M.1 | Maltitol | Unstandardized foods | Good Manufacturing Practice |
| M.2 | Maltitol syrup | Unstandardized foods | Good Manufacturing Practice |
| M.3 | Mannitol | Unstandardized foods | Good Manufacturing Practice |
| N.1 | Neotame | (1) Table-top sweeteners | (1) Good Manufacturing Practice |
| (2) Breakfast cereals | (2) 0.016% | ||
| (3) Unstandardized beverage concentrates; Unstandardized beverage mixes; Unstandardized beverages | (3) 0.003% in beverages as consumed | ||
| (4) Filling mixes; Fillings; Topping mixes; Toppings; Unstandardized dessert mixes; Unstandardized desserts; Yogurt | (4) 0.01% in products as consumed | ||
| (5) Breath freshener products; Chewing gum | (5) 0.032% | ||
| (6) Unstandardized fruit spreads; Unstandardized purées; Unstandardized sauces; Unstandardized table syrups | (6) 0.007% | ||
| (7) Nut spreads; Peanut spreads; Unstandardized salad dressings | (7) 0.002% | ||
| (8) Unstandardized condiments | (8) 0.007% | ||
| (9) Confectionery glazes for snack foods; Sweetened seasonings or coating mixes for snack foods | (9) 0.0032% | ||
| (10) Unstandardized confectionery; Unstandardized confectionery coatings | (10) 0.01% | ||
| (11) Baking mixes; Unstandardized bakery products | (11) 0.013% in products as consumed | ||
| S.1 | Sorbitol | (1) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (1) 6.0% |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.1.1 | Sorbitol syrup | Unstandardized foods | Good Manufacturing Practice |
| S.2 | Sucralose | (1) Table-top sweeteners | (1) Good Manufacturing Practice |
| (2) Breakfast cereals | (2) 0.1% | ||
| (3) Unstandardized beverage concentrates; Unstandardized beverage mixes; Unstandardized beverages; Unstandardized dairy beverages | (3) 0.025% in beverages as consumed | ||
| (4) Filling mixes; Fillings; Topping mixes; Toppings; Unstandardized dessert mixes; Unstandardized desserts; Yogurt | (4) 0.025% in products as consumed | ||
| (5) Chewing gum; Breath freshener products | (5) 0.15% | ||
| (6) Unstandardized fruit spreads | (6) 0.045% | ||
| (7) Unstandardized condiments; Unstandardized salad dressings | (7) 0.04% | ||
| (8) Confectionery glazes for snack foods; Sweetened seasonings or coating mixes for snack foods; Unstandardized confectionery; Unstandardized confectionery coatings | (8) 0.07% | ||
| (9) Baking mixes; Unstandardized bakery products | (9) 0.065% in products as consumed | ||
| (10) Unstandardized processed fruit and vegetable products, except unstandardized canned fruit | (10) 0.015% | ||
| (11) Unstandardized alcoholic beverages | (11) 0.07% | ||
| (12) Puddings; Pudding mixes | (12) 0.04% in products as consumed | ||
| (13) Unstandardized table syrups | (13) 0.15% | ||
| (14) Canned (naming the fruit); Unstandardized canned fruit | (14) 0.025% | ||
| (15) Pickles; Relishes | (15) 0.015% | ||
| T.1 | Thaumatin | (1) Chewing gum; Breath freshener products | (1) 500 p.p.m. |
| (2) Salt substitutes | (2) 400 p.p.m. | ||
| (3) (naming the flavour) Flavour referred to in section B.10.005; Unstandardized flavouring preparations | (3) 100 p.p.m. | ||
| X.1 | Xylitol | Unstandardized foods | Good Manufacturing Practice |
- SOR/93-276, s. 12
- SOR/94-625, s. 5
- SOR/94-689, s. 2(F)
- SOR/94-779, s. 3
- SOR/97-512, ss. 3, 4
- SOR/2004-261, s. 2
- SOR/2007-76, s. 3
- SOR/2007-176, s. 7
- SOR/2010-142, ss. 52 to 55
- SOR/2012-104, ss. 11 to 15
TABLE X
Food Additives That May Be Used as pH Adjusting Agents, Acid-Reacting Materials and Water Correcting Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acetic Acid | (1) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (1) Good Manufacturing Practice |
| (2) Canned Asparagus | (2) Good Manufacturing Practice | ||
| (3) Gelatin | (3) Good Manufacturing Practice | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| A.2 | Adipic Acid | Unstandardized foods | Good Manufacturing Practice |
| A.3 | Ammonium Aluminum Sulphate | (1) Baking powder | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.4 | Ammonium Bicarbonate | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.5 | Ammonium Carbonate | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.6 | Ammonium Citrate, dibasic | Unstandardized foods | Good Manufacturing Practice |
| A.7 | Ammonium Citrate, monobasic | Unstandardized foods | Good Manufacturing Practice |
| A.8 | Ammonium Hydroxide | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Gelatin | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| A.9 | Ammonium Phosphate, dibasic | (1) Ale; Bacterial cultures; Baking powder; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| A.10 | Ammonium Phosphate, monobasic | (1) Ale; Bacterial cultures; Baking powder; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| (3) Uncultured buttermilk | (3) 0.1% | ||
| C.1 | Calcium Acetate | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.1A | Calcium Acid Pyrophosphate | (1) Baking powder | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.2 | Calcium Carbonate | (1) Ice cream mix; Ice milk mix; Wine | (1) Good Manufacturing Practice |
| (2) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (2) Good Manufacturing Practice | ||
| (3) Grape juice | (3) Good Manufacturing Practice | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Cocoa products | (5) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| C.3 | Calcium Chloride | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.4 | Calcium Citrate | (1) Infant formula | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.5 | Calcium Fumarate | Unstandardized foods | Good Manufacturing Practice |
| C.6 | Calcium Gluconate | Unstandardized foods | Good Manufacturing Practice |
| C.7 | Calcium Hydroxide | (1) Ale; Beer; Ice cream mix; Ice milk mix; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Canned peas | (2) 0.01% | ||
| (3) Infant formula | (3) Good Manufacturing Practice | ||
| (4) Grape Juice | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| C.8 | Calcium Lactate | (1) Baking powder | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.9 | Calcium Oxide | (1) Ale; Beer; Ice cream mix; Ice milk mix; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.10 | Calcium Phosphate, dibasic | Unstandardized foods | Good Manufacturing Practice |
| C.11 | Calcium Phosphate, monobasic | (1) Ale; Baking powder; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| C.12 | Calcium Phosphate, tribasic | Unstandardized foods | Good Manufacturing Practice |
| C.13 | Calcium Sulphate | Ale; Beer; Light beer; Malt liquor; Porter; Stout; Wine | Good Manufacturing Practice |
| C.13A | Carbon Dioxide | (Naming the variety) Cheese | Good Manufacturing Practice |
| C.14 | Citric Acid | (1) Ale; Beer; Cider; Honey wine; Malt liquor; Porter; Stout; Wine | (1) Good Manufacturing Practice |
| (2) Apple (or rhubarb) and (naming the fruit) jam; Apricot nectar; Beans; Beans with pork; Canned mushrooms; Canned (naming the fruit); Canned (naming the vegetable); Canned tomatoes; Concentrated tomato paste; Fig marmalade; Fig marmalade with pectin; Frozen mushrooms; Frozen (naming the fruit); Frozen (naming the vegetable); Grape juice; Mincemeat; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; Olives; Peach nectar; Pear nectar; Pickles; Pineapple marmalade; Pineapple marmalade with pectin; Relishes; Tomato juice; Tomato paste; Tomato pulp; Tomato puree | (2) Good Manufacturing Practice | ||
| (3) Canned shellfish; Canned spring mackerel; Dried egg-white (dried albumen); Dried whole egg; Dried yolk; Frozen cooked shrimp; Frozen egg-white (frozen albumen); Frozen whole egg; Frozen yolk; Gelatin; Liquid egg-white (liquid albumen); Liquid whole egg; Liquid yolk | (3) Good Manufacturing Practice | ||
| (4) Cocoa products | (4) 1.0%, singly or in combination with tartaric acid, calculated on a fat-free basis | ||
| (5) Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cottage cheese; Cream cheese spread; Cream cheese spread with (naming the added ingredients); Creamed cottage cheese; Ice cream mix; Ice milk mix; (naming the variety) Whey cheese; Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Sherbet | (5) Good Manufacturing Practice | ||
| (6) French dressing; Margarine; Mayonnaise; Salad dressing | (6) Good Manufacturing Practice | ||
| (7) Infant formula | (7) Good Manufacturing Practice | ||
| (8) Unstandardized foods | (8) Good Manufacturing Practice | ||
| C.15 | Cream of Tartar | Same foods as listed for Potassium Acid Tartrate | Same levels as prescribed for Potassium Acid Tartrate |
| F.1 | Fumaric Acid | (1) Gelatin | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Wine | (3) Good Manufacturing Practice | ||
| G.1 | Gluconic Acid | Unstandardized foods | Good Manufacturing Practice |
| G.2 | Glucono-delta-lactone | Unstandardized foods | Good Manufacturing Practice |
| H.1 | Hydrochloric Acid | (1) Ale; Beer; Gelatin; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Infant formula | (2) Good Manufacturing Practice | ||
| L.1 | Lactic Acid | (1) Ale; Baking powder; Beer; Bread; Cider; Cottage cheese; Creamed cottage cheese; Dried egg-white (dried albumen); Dried whole egg; Dried yolk; French dressing; Frozen egg-white (frozen albumen); Frozen whole egg; Frozen yolk; Ice cream mix; Ice milk mix; Liquid egg-white (liquid albumen); Liquid whole egg; Liquid yolk; Malt liquor; Mayonnaise; Olives; Pickles; Porter; Relishes; Salad dressing; Sherbet; Stout | (1) Good Manufacturing Practice |
| (2) Canned pears; Canned strawberries | (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) Good Manufacturing Practice | ||
| (4) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Wine | (6) Good Manufacturing Practice | ||
| M.2 | Magnesium Carbonate | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Ice cream mix; Ice milk mix | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| M.3 | Magnesium Citrate | Soft drinks | Good Manufacturing Practice |
| M.4 | Magnesium Fumarate | Unstandardized foods | Good Manufacturing Practice |
| M.5 | Magnesium Hydroxide | (1) Canned peas | (1) 0.05% |
| (2) Cocoa products | (2) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| (3) Gelatin; Ice cream mix; Ice milk mix | (3) Good Manufacturing Practice | ||
| (4) Bacterial cultures | (4) Good Manufacturing Practice | ||
| M.6 | Magnesium Oxide | Ice cream mix; Ice milk mix | Good Manufacturing Practice |
| M.6A | Magnesium Phosphate | Bacterial cultures | Good Manufacturing Practice |
| M.7 | Magnesium Sulphate | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bacterial cultures | (2) Good Manufacturing Practice | ||
| M.8 | Malic Acid | (1) Apple (or rhubarb) and (naming the fruit) jam; Apricot nectar; Canned asparagus; Fig marmalade; Fig marmalade with pectin; Frozen (naming the fruit); (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Peach nectar; Pear nectar; Pineapple marmalade; Pineapple marmalade with pectin | (1) Good Manufacturing Practice |
| (2) Canned applesauce; Canned pears; Canned strawberries | (2) Good Manufacturing Practice | ||
| (3) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (3) Good Manufacturing Practice | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Wine | (5) Good Manufacturing Practice | ||
| M.8A | Manganese Sulphate | Bacterial cultures | Good Manufacturing Practice |
| M.9 | Metatartaric Acid | Wine | 0.01% |
| P.1 | Phosphoric Acid | (1) Ale; Beer; Cottage Cheese; Creamed cottage cheese; Gelatin; Light beer; Malt liquor; Mono-glycerides and mono- and diglycerides; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (2) Good Manufacturing Practice | ||
| (3) Fish protein | (3) Good Manufacturing Practice | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Cocoa products | (5) 0.5%, expressed as P2O5, calculated on a fat-free basis | ||
| P.2 | Potassium Acid Tartrate | (1) Baking powder; Honey wine | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Wine | (3) 0.42% | ||
| P.3 | Potassium Aluminum Sulphate | (1) Ale; Baking powder; Beer; Light beer; Malt liquor; Oil-soluble annatto; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| P.4 | Potassium Bicarbonate | (1) Baking powder; Malted milk; Malted milk powder | (1) Good Manufacturing Practice |
| (2) Cocoa products | (2) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| (3) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (3) Good Manufacturing Practice | ||
| (4) Infant formula | (4) Good Manufacturing Practice | ||
| (5) Margarine | (5) Good Manufacturing Practice | ||
| (6) Unstandardized foods | (6) Good Manufacturing Practice | ||
| (7) Wine | (7) Good Manufacturing Practice | ||
| P.5 | Potassium Carbonate | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) Good Manufacturing Practice | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) A blend of prepared fish and prepared meat referred to in paragraph B.21.006(n) | (5) Good Manufacturing Practice | ||
| (6) Wine | (6) Good Manufacturing Practice | ||
| P.6 | Potassium Chloride | Ale; Beer; Light beer; Malt liquor; Porter; Stout | Good Manufacturing Practice |
| P.7 | Potassium Citrate | (1) Infant formula | (1) Good Manufacturing Practice |
| (2) Margarine | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| (4) Wine | (4) Good Manufacturing Practice | ||
| P.8 | Potassium Fumarate | Unstandardized foods | Good Manufacturing Practice |
| P.9 | Potassium Hydroxide | (1) Oil-soluble annatto | (1) 1.0% |
| (2) Cocoa products | (2) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| (3) Ice cream mix; Ice milk mix; Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product | (3) Good Manufacturing Practice | ||
| (4) Infant formula | (4) Good Manufacturing Practice | ||
| (5) Margarine | (5) Good Manufacturing Practice | ||
| (6) Grape juice | (6) Good Manufacturing Practice | ||
| (7) Unstandardized foods | (7) Good Manufacturing Practice | ||
| P.9A | Potassium Lactate | Margarine | Good Manufacturing Practice |
| P.10 | Potassium Phosphate, dibasic | Unstandardized foods | Good Manufacturing Practice |
| P.11 | Potassium Sulphate | Ale; Beer; Light beer; Malt liquor; Porter; Stout | Good Manufacturing Practice |
| P.12 | Potassium Tartrate | Cider | Good Manufacturing Practice |
| S.1 | Sodium Acetate | Unstandardized foods | Good Manufacturing Practice |
| S.2 | Sodium Acid Pyrophosphate | (1) Baking powder | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.3 | Sodium Acid Tartrate | Baking Powder | Good Manufacturing Practice |
| S.4 | Sodium Aluminum Phosphate | Unstandardized foods | Good Manufacturing Practice |
| S.5 | Sodium Aluminum Sulphate | (1) Baking powder | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.6 | Sodium Bicarbonate | (1) Apple (or rhubarb) and (naming the fruit) jam; Baking powder; Dried egg-white (dried albumen); Dried whole egg; Dried yolk; Fig marmalade; Fig marmalade with pectin; Frozen egg-white (frozen albumen); Frozen whole egg; Frozen yolk; Ice cream mix; Ice milk mix; Liquid egg-white (liquid albumen); Liquid whole egg; Liquid yolk; Malted milk powder; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; Oil-soluble annatto; Pineapple marmalade; Pineapple marmalade with pectin; Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product | (1) Good Manufacturing Practice |
| (2) Cocoa products | (2) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| (3) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (3) Good Manufacturing Practice | ||
| (4) Infant formula | (4) Good Manufacturing Practice | ||
| (5) Margarine | (5) Good Manufacturing Practice | ||
| (6) Unstandardized foods | (6) Good Manufacturing Practice | ||
| S.7 | Sodium Bisulphate | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| S.8 | Sodium Carbonate | (1) Apple (or rhubarb) and (naming the fruit) jam; Dried egg-white (dried albumen); Dried whole egg; Dried yolk; Fig marmalade; Fig marmalade with pectin; Frozen egg-white (frozen albumen); Frozen whole egg; Frozen yolk; Gelatin; Ice cream mix; Ice milk mix; Liquid egg-white (liquid albumen); Liquid whole egg; Liquid yolk; Meat binder or (naming the meat product) binder where sold for use in preserved meat or preserved meat by-product; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; Pineapple marmalade; Pineapple marmalade with pectin | (1) Good Manufacturing Practice |
| (2) Cocoa products | (2) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 | ||
| (3) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (3) Good Manufacturing Practice | ||
| (4) Margarine | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| S.9 | Sodium Citrate | (1) Apple (or rhubarb) and (naming the fruit) jam; Cottage cheese; Cream; Creamed cottage cheese; Ice cream mix; Ice milk mix; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Pineapple marmalade or Fig marmalade; Pineapple marmalade with pectin or Fig marmalade with pectin; Sherbet | (1) Good Manufacturing Practice |
| (2) Infant formula | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| (4) Margarine | (4) Good Manufacturing Practice | ||
| S.12 | Sodium Fumarate | Unstandardized foods | Good Manufacturing Practice |
| S.13 | Sodium Gluconate | Unstandardized foods | Good Manufacturing Practice |
| S.14 | Sodium Hexametaphosphate | Unstandardized foods | Good Manufacturing Practice |
| S.15 | Sodium Hydroxide | (1) Cocoa products | (1) Sufficient to process the cocoa products in accordance with the requirements of section B.04.005 |
| (2) Gelatin; Ice cream mix; Ice milk mix; (naming the flavour) Partly skimmed milk; (naming the flavour) Skim milk; Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product | (2) Good Manufacturing Practice | ||
| (3) Infant formula | (3) Good Manufacturing Practice | ||
| (4) Margarine | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) (Naming the variety) Whey cheese; Whey cheese | (6) Good Manufacturing Practice | ||
| S.16 | Sodium Lactate | (1) Margarine | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.17 | Sodium Phosphate, dibasic | (1) Ale; Bacterial culture; Beer; Cream; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.18 | Sodium Phosphate, monobasic | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.19 | Sodium Phosphate, tribasic | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.20 | Sodium Potassium Tartrate | (1) Apple (or rhubarb) and (naming the fruit) jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Pineapple marmalade or Fig marmalade; Pineapple marmalade with pectin or Fig marmalade with pectin | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) Good Manufacturing Practice | ||
| S.21 | Sodium Pyrophosphate, tetrabasic | Unstandardized foods | Good Manufacturing Practice |
| S.22 | Sodium Tripolyphosphate | Unstandardized foods | Good Manufacturing Practice |
| S.23 | Sulphuric Acid | Ale; Beer; Light beer; Malt liquor; Porter; Stout | Good Manufacturing Practice |
| S.24 | Sulphurous Acid | Gelatin | Good Manufacturing Practice provided the finished product does not contain more than 500 p.p.m. calculated as sulphur dioxide |
| T.1 | Tartaric Acid | (1) Ale; Apple (or rhubarb) and (naming the fruit) jam; Baking powder; Beer; Cider; Canned asparagus; Fig marmalade; Fig marmalade with pectin; French dressing; Honey wine; Ice cream mix; Ice milk mix; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly; (naming the fruit) Jelly with pectin; Light beer; Malt liquor; (naming the citrus fruit) Marmalade; (naming the citrus fruit) Marmalade with pectin; Mayonnaise; Pineapple marmalade; Pineapple marmalade with pectin; Porter; Salad dressing; Sherbet; Stout; Wine | (1) Good Manufacturing Practice |
| (2) Canned pears; Canned strawberries | (2) Good Manufacturing Practice | ||
| (3) Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients); (naming the variety) Whey cheese | (3) Good Manufacturing Practice | ||
| (4) Margarine | (4) Good Manufacturing Practice | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Cocoa products | (6) 1%, singly or in combination with citric acid, calculated on a fat-free basis |
- SOR/78-874, s. 4
- SOR/79-660, ss. 14 to 17
- SOR/79-664, ss. 3 to 13
- SOR/79-752, s. 9
- SOR/80-501, s. 4
- SOR/86-1112, ss. 6 to 8
- SOR/92-106, s. 1
- SOR/92-344, s. 5
- SOR/94-689, s. 2(F)
- SOR/95-281, ss. 2 to 5
- SOR/95-436, ss. 2, 3
- SOR/97-263, ss. 11 to 25
- SOR/97-561, s. 3
- SOR/98-580, s. 1(F)
- SOR/2001-94, s. 3
- SOR/2006-91, ss. 13 to 20
- SOR/2007-75, s. 8
- SOR/2010-41, s. 9(E)
- SOR/2010-94, s. 8(E)
- SOR/2010-142, s. 56
- SOR/2010-143, ss. 32 to 36
- SOR/2012-43, ss. 32 to 34, 35(F), 36
- SOR/2012-103, ss. 1(F), 2
TABLE XI
PART I
Food Additives That May Be Used as Class I Preservatives
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acetic Acid | (1) Preserved fish; Preserved meat; Preserved meat by-product; Preserved poultry meat; Preserved poultry meat by-product; Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.2 | Ascorbic Acid | (1) Ale; Beer; Canned mushrooms; Canned tuna; Canned white asparagus; Cider; Frozen fruit; Glaze of Frozen fish; Headcheese; Light beer; Malt liquor; Meat binder for preserved meat and preserved meat by-product (Division 14 only); Porter; Preserved fish; Frozen minced fish; Frozen comminuted fish; Preserved meat; Preserved meat by-product; Preserved poultry meat; Preserved poultry meat by-product; Pumping pickle; Cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product; Stout; Wine | (1) Good Manufacturing Practice |
| (2) Canned applesauce | (2) If used either singly or in combination with Iso-Ascorbic Acid, the total not to exceed 150 p.p.m. | ||
| (3) Canned peaches | (3) 550 p.p.m. | ||
| (4) Unstandardized foods | (4) Good Manufacturing practice | ||
| (5) Canned mandarin oranges | (5) 400 p.p.m. | ||
| C.1 | Calcium Ascorbate | Same foods as listed for Ascorbic Acid | Same levels as prescribed for Ascorbic Acid |
| E.1 | Erythorbic Acid | (1) Ale; Beer; Cider; Frozen fruit; Headcheese; Light beer; Malt liquor; Meat binder for preserved meat and preserved meat by-product (Division 14 only); Porter; Preserved fish; Frozen minced fish; Frozen comminuted fish; Glaze of frozen fish; Preserved meat; Preserved meat by-product; Preserved poultry meat; Preserved poultry meat by-product; Pumping pickle; Cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product; Stout; Wine | (1) Good Manufacturing Practice |
| (2) Canned applesauce | (2) If used either singly or in combination with Ascorbic Acid, the total not to exceed 150 p.p.m. | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| I.1 | Iso-Ascorbic Acid | Same foods as listed for Erythorbic Acid | Same levels as prescribed for Erythorbic Acid |
| P.1 | Potassium Nitrate | (1) Meat binder for dry sausage, semi-dry sausage, preserved meat and preserved meat by-products prepared by slow cure processes (Division 14) | (1) When the meat binder is used in accordance with label instructions, whether potassium nitrate is added alone or in combination with sodium nitrate, the total amount of such nitrates thereby added to each batch of dry sausage, semi-dry sausage, preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation |
| (2) Cover pickle and dry cure employed in the curing of preserved meat and preserved meat by-products prepared by slow cure processes (Division 14) | (2) When the cover pickle or dry cure is used in accordance with label instructions, whether potassium nitrate is added alone or in combination with sodium nitrate, the total amount of such nitrates thereby added to each batch of preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (3) Dry sausage, semi-dry sausage, preserved meat and preserved meat by-products prepared by slow cure processes (Division 14) | (3) Where potassium nitrate is added alone or in combination with sodium nitrate, the total amount of such nitrates added to each batch of dry sausage, semi-dry sausage, preserved meat or preserved meat by-products shall not exceed 0.32 ounces per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (4) Ripened cheese, containing not more than 68% moisture on a fat free basis during manufacture of which the lactic acid fermentation and salting is completed later than 12 hours after coagulation of the curd by food enzymes and where the added salt is applied externally to the cheese as dry salt or in the form of brine | (4) If used singly or in combination with sodium nitrate, the total not to exceed 200 p.p.m. (based in milk). Residue in the finished cheese not to exceed 50 p.p.m. | ||
| (5) Mold ripened cheese packed in hermetically sealed containers | (5) If used singly or in combination with sodium nitrate, the total not to exceed 200 p.p.m. (based in milk). Residue in the finished cheese not to exceed 50 p.p.m. | ||
| P.2 | Potassium Nitrite | (1) Meat binder, pumping pickle, cover pickle and dry cure employed in the curing of preserved meat and preserved meat by-products (Division 14) | (1) When the meat binder, pumping pickle, cover pickle or dry cure is used in accordance with label instructions, whether potassium nitrite is added alone or in combination with sodium nitrite, the total amount of such nitrites thereby added to each batch of preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million calculated prior to any smoking, cooking or fermentation |
| (2) Preserved meat except side bacon and preserved meat by-products (Division 14) | (2) Where potassium nitrite is added alone or in combination with sodium nitrite, the total amount of such nitrites added to each batch of preserved meat, except side bacon or preserved meat by-products, shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (3) Side bacon | (3) Where potassium nitrite is added alone or in combination with sodium nitrite, the total amount of such nitrites added to each batch of side bacon shall not exceed 0.19 ounce per 100 pounds or 120 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (4) Preserved poultry meat and preserved poultry meat by-products (Division 22) | (4) Where potassium nitrite is added alone or in combination with sodium nitrite, the total amount of such nitrites added to each batch of preserved poultry meat or preserved poultry meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| S.1 | Sodium Ascorbate | Same foods as listed for Ascorbic Acid | Same levels as prescribed for Ascorbic Acid |
| S.2 | Sodium Erythorbate | (1) Same foods as listed for Erythorbic Acid | (1) Same levels as prescribed for Erythorbic Acid |
| (2) Canned clams | (2) 350 p.p.m. | ||
| S.3 | Sodium Iso-Ascorbate | Same foods as listed for Erythorbic Acid | Same levels as prescribed for Erythorbic Acid |
| S.4 | Sodium Nitrate | (1) Meat binder for dry sausage, semi-dry sausage, preserved meat and preserved meat by-products prepared by slow cure processes (Division 14) | (1) When the meat binder is used in accordance with label instructions, whether sodium nitrate is added alone or in combination with potassium nitrate, the total amount of such nitrates thereby added to each batch of dry sausage, semi-dry sausage, preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation |
| (2) Cover pickle and dry cure employed in the curing of preserved meat and preserved meat by-products prepared by slow cure processes (Division 14) | (2) When the cover pickle or dry cure is used in accordance with label instructions, whether sodium nitrate is added alone or in combination with potassium nitrate, the total amount of such nitrates thereby added to each batch of preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (3) Dry sausage, semi-dry sausage, preserved meat and preserved meat by-products prepared by the slow cure processes (Division 14) | (3) Where sodium nitrate is added alone or in combination with potassium nitrate, the total amount of such nitrates added to each batch of dry sausage, semi-dry sausage, preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (4) Ripened cheese, containing not more than 68% moisture on a fat free basis during manufacture of which the lactic acid fermentation and salting is completed later than 12 hours after coagulation of the curd by food enzymes and where the added salt is applied externally to the cheese as dry salt or in the form of brine | (4) If used singly or in combination with potassium nitrate, the total not to exceed 200 p.p.m. (based in milk). Residue in the finished cheese not to exceed 50 p.p.m. | ||
| (5) Mold ripened cheese packed in hermetically sealed containers | (5) If used singly or in combination with potassium nitrate, the total not to exceed 200 p.p.m. (based in milk). Residue in the finished cheese not to exceed 50 p.p.m. | ||
| S.5 | Sodium Nitrite | (1) Meat binder, pumping pickle, cover pickle and dry cure employed in the curing of preserved meat and preserved meat by-products (Division 14) | (1) When the meat binder, pumping pickle, cover pickle or dry cure is used in accordance with label instructions, whether sodium nitrite is added alone or in combination with potassium nitrite, the total amount of such nitrites thereby added to each batch of preserved meat or preserved meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation |
| (2) Preserved meat, except side bacon, and preserved meat by-products (Division 14) | (2) Where sodium nitrite is added alone or in combination with potassium nitrite, the total amount of such nitrites added to each batch of preserved meat, except side bacon or preserved meat by-products, shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (3) Side bacon | (3) Where sodium nitrite is added alone or in combination with potassium nitrite, the total amount of such nitrites added to each batch of side bacon shall not exceed 0.19 ounce per 100 pounds or 120 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| (4) Preserved poultry meat and preserved poultry meat by-products (Division 22) | (4) Where sodium nitrite is added alone or in combination with potassium nitrite, the total amount of such nitrites added to each batch of preserved poultry meat or preserved poultry meat by-products shall not exceed 0.32 ounce per 100 pounds or 200 parts per million, calculated prior to any smoking, cooking or fermentation | ||
| W.1 | Wood Smoke | (1) (naming the variety) Cheese; Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (1) Good Manufacturing Practice |
| (2) Preserved fish; Preserved meat (Divisions 14 and 21); Preserved meat by-product (Divisions 14 and 21); Preserved poultry meat; Preserved poultry meat by-product; Sausage | (2) Good Manufacturing Practice | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice |
PART II
Food Additives That May Be Used as Class II Preservatives
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| B.1 | Benzoic Acid | (1) Apple (or rhubarb) and (naming the fruit) jam; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Mincemeat; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; (naming the fruit) Juice; (naming the fruits) Juice; Packaged fish and meat products that are marinated or otherwise cold-processed (Division 21); Pickles; Pineapple marmalade with pectin; Relishes; Tomato catsup; Tomato paste; Tomato pulp; Tomato puree | (1) 1,000 p.p.m. |
| (2) 1,000 p.p.m. | ||
| (3) Margarine | (3) If used singly or in combination with Sorbic Acid, the total shall not exceed 1,000 p.p.m. | ||
| C.1 | Calcium Sorbate | Same foods as listed for sorbic acid | Same levels as prescribed for Sorbic Acid |
| C.2 | Carnobacterium maltaromaticum CB1 | (1) Vacuum-packed wieners | (1) Good Manufacturing Practice |
| (2) Vacuum-packed sliced roast beef in accordance with section B.14.005 | (2) Good Manufacturing Practice | ||
| (3) Vacuum-packed sliced cooked ham in accordance with section B.14.005 or B.14.031 | (3) Good Manufacturing Practice | ||
| (4) Vacuum-packed sliced cooked turkey in accordance with section B.22.006 or B.22.021 | (4) Good Manufacturing Practice | ||
| H.1 | 4-Hexylresorcinol | Crustaceans | Good Manufacturing Practice. Residues in the edible portion of the uncooked product not to exceed 1.0 p.p.m. |
| M.1 | Methyl-ρ-hydroxy Benzoate | (1) Apple (or rhubarb) and (naming the fruit) jam; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Mincemeat; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; (naming the fruit) Juice; (naming the fruits) Juice; Packaged fish and meat products that are marinated or otherwise cold-processed (Division 21); Pickles; Pineapple marmalade with pectin; Relishes; Tomato catsup; Tomato paste; Tomato pulp; Tomato puree | (1) 1,000 p.p.m. |
| (2) 1,000 p.p.m. | ||
| M.2 | Methyl Paraben | Same foods as listed for Methyl-p-hydroxy Benzoate | Same levels as prescribed for Methyl-p-hydroxy Benzoate |
| P.1 | Potassium Benzoate | Same foods as listed for Benzoic Acid | 1,000 p.p.m. calculated as Benzoic Acid |
| P.2 | Potassium Bisulphite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| P.3 | Potassium Metabisulphite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| P.4 | Potassium Sorbate | Same foods as listed for Sorbic Acid | Same levels as prescribed for Sorbic Acid |
| P.5 | Propyl-ρ-hydroxy Benzoate | (1) Apple (or rhubarb) and (naming the fruit) jam; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Mincemeat; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; (naming the fruit) Juice; (naming the fruits) Juice; Packaged fish and meat products that are marinated or otherwise cold-processed (Division 21); Pickles; Pineapple marmalade with pectin; Relishes; Tomato catsup; Tomato paste; Tomato pulp; Tomato puree | (1) 1,000 p.p.m. |
| (2) 1,000 p.p.m. | ||
| P.6 | Propyl Paraben | Same foods as listed for Propyl-p-hydroxy Benzoate | Same levels as prescribed for Propyl-p-hydroxy Benzoate |
| S.01 | Sodium Acetate | (1) Brawn; Headcheese; Meat by-product loaf; Meat loaf; Potted meat; Potted meat by-product; Prepared meat; Prepared meat by-product; Prepared poultry meat; Prepared poultry meat by-product; Preserved meat; Preserved meat by-product; Preserved poultry meat; Preserved poultry meat by-product; Sausage | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| S.1 | Sodium Benzoate | Same foods as listed for Benzoic Acid | 1,000 p.p.m. calculated as Benzoic Acid |
| S.2 | Sodium Bisulphite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| S.21 | Sodium Diacetate | (1) Brawn; Headcheese; Meat by-product loaf; Meat loaf; Potted meat; Potted meat by-product; Prepared fish or prepared meat (Division 21); Prepared meat; Prepared meat by-product; Prepared poultry meat; Prepared poultry meat by-product; Preserved fish or preserved meat (Division 21); Preserved meat; Preserved meat by-product; Preserved poultry meat; Preserved poultry meat by-product; Sausage | (1) 0.25% of final product weight |
| (2) 0.25% of final product weight | ||
| S.3 | Sodium Metabisulphite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| S.4 | Sodium Salt of Methyl-ρ-hydroxy Benzoic Acid | Same foods as listed for Methyl-p-hydroxy Benzoate | 1,000 p.p.m. calculated as Methyl-p-hydroxy Benzoate |
| S.5 | Sodium Salt of Propyl-ρ-hydroxy Benzoic Acid | Same foods as listed for Propyl-p-hydroxy Benzoate | 1,000 p.p.m. calculated as Propyl-p-hydroxy Benzoate |
| S.6 | Sodium Sorbate | Same foods as listed for Sorbic Acid | Same levels as prescribed for Sorbic Acid |
| S.7 | Sodium Sulphite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| S.8 | Sodium Dithionite | Same foods as listed for Sulphurous Acid | Same levels as prescribed for Sulphurous Acid |
| S.9 | Sorbic Acid | (1) Apple (or rhubarb) and (naming the fruit) jam; Cold-processed smoked and salted fish paste; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fig marmalade with pectin; Mincemeat; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; (naming the fruit) Juice; (naming the fruits) Juice; (naming the source of the glucose) Syrup; Pickles; Pineapple marmalade with pectin; Relishes; Smoked or salted dried fish; Tomato catsup; Tomato paste; Tomato pulp; Tomato puree | (1) 1,000 p.p.m. |
| (2) 1,000 p.p.m. | ||
| (3) Olive brine | (3) 300 p.p.m. | ||
| (4) Margarine | (4) If used singly or in combination with Benzoic Acid, the total shall not exceed 1,000 p.p.m. | ||
| (5) Unstandardized salad dressings | (5) 3,350 p.p.m. | ||
| S.10 | Sulphurous Acid | (1) Cider; Honey wine; Wine | (1) 70 p.p.m. in the free state or 350 p.p.m. in the combined state calculated as sulphur dioxide |
| (2) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (2) 15 p.p.m. calculated as sulphur dioxide | ||
| (3) Apple (or rhubarb) and (naming the fruit) jam; Concentrated (naming the fruit) juice except frozen concentrated orange juice; Fancy molasses; Fig marmalade with pectin; Frozen sliced apples; Gelatin; Mincemeat; (naming the citrus fruit) Marmalade with pectin; (naming the fruit) Jam; (naming the fruit) Jam with pectin; (naming the fruit) Jelly with pectin; (naming the fruit) Juice; (naming the fruits) Juice; (naming the source of the glucose) Syrup; Pickles; Pineapple marmalade with pectin; Refiners’ molasses; Relishes; Table molasses; Tomato catsup; Tomato paste; Tomato pulp; Tomato puree | (3) 500 p.p.m. calculated as sulphur dioxide | ||
| (4) Unstandardized beverages | (4) 100 p.p.m. calculated as sulphur dioxide | ||
| (5) Dried fruits and vegetables | (5) 2,500 p.p.m. calculated as sulphur dioxide | ||
| (6) 500 p.p.m. calculated as sulphur dioxide | ||
| (7) Frozen mushrooms | (7) 90 p.p.m. calculated as sulphur dioxide | ||
| (8) Dextrose Anhydrous; Dextrose Monohydrate | (8) 20 p.p.m. calculated as sulphur dioxide | ||
| (9) Glucose or glucose syrup | (9) 40 p.p.m. except glucose or glucose syrup for the manufacture of sugar confectionery not more than 400 p.p.m. calculated as sulphur dioxide | ||
| (10) Glucose solids or dried glucose syrup | (10) 40 p.p.m. except glucose solids or dried glucose syrup for the manufacture of sugar confectionary not more than 150 p.p.m. calculated as sulphur dioxide | ||
| (11) Crustaceans | (11) Good Manufacturing Practice. Residues in the edible portion of the uncooked product not to exceed 100 p.p.m., calculated as sulphur dioxide. |
PART III
Food Additives That May Be Used as Class III Preservatives
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| C.1 | Calcium Propionate | (1) Same foods as listed for Propionic Acid | (1) 2,000 p.p.m. calculated as Propionic Acid |
| (2) Soft flour tortillas | (2) 4,000 p.p.m. | ||
| C.2 | Calcium Sorbate | Same foods as listed for Sorbic Acid | Same maximum levels of use as listed for Sorbic Acid |
| N.1 | Natamycin | (1) The surface of (naming the variety) cheese and cheddar cheese | (1) 20 p.p.m. in accordance with the requirements of sections B.08.033 and B.08.034 |
| (2) The surface of grated or shredded (naming the variety) cheese and grated or shredded cheddar cheese | (2) 10 p.p.m. in accordance with the requirements of sections B.08.033 and B.08.034 | ||
| P.1 | Potassium Sorbate | (1) Same foods as listed for Sorbic Acid | (1) Same maximum levels of use as listed for Sorbic Acid |
| (2) Soft flour tortillas | (2) 5,000 p.p.m. | ||
| P.2 | Propionic Acid | (1) Bread | (1) 2,000 p.p.m. |
| (2) (naming the variety) Cheese; Cheddar cheese; Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (2) 2,000 p.p.m. or 3,000 p.p.m., as the case may be, in accordance with the requirements of sections B.08.033, B.08.034, B.08.035, B.08.037, B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3, B.08.041.4, B.08.041.5, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (3) 2,000 p.p.m. | ||
| S.1 | Sodium Diacetate | (1) Bread | (1) 3,000 p.p.m. |
| (2) 3,000 p.p.m. | ||
| S.2 | Sodium Propionate | Same foods as listed for Propionic Acid | 2,000 p.p.m. calculated as Propionic Acid |
| S.3 | Sodium Sorbate | Same foods as listed for Sorbic Acid | Same maximum levels of use as listed for Sorbic Acid |
| S.4 | Sorbic Acid | (1) Bread | (1) 1,000 p.p.m. |
| (2) (naming the variety) Cheese; Cheddar cheese; Cream cheese; Cream cheese with (naming the added ingredients); Cream cheese spread; Cream cheese spread with (naming the added ingredients); Processed (naming the variety) cheese; Processed (naming the variety) cheese with (naming the added ingredients); Processed cheese food; Processed cheese food with (naming the added ingredients); Processed cheese spread; Processed cheese spread with (naming the added ingredients); Cold-pack (naming the variety) cheese; Cold-pack (naming the variety) cheese with (naming the added ingredients); Cold-pack cheese food; Cold-pack cheese food with (naming the added ingredients) | (2) 3,000 p.p.m. in accordance with the requirements of sections B.08.033, B.08.034, B.08.035, B.08.037, B.08.038, B.08.039, B.08.040, B.08.041, B.08.041.1, B.08.041.2, B.08.041.3, B.08.041.4, B.08.041.5, B.08.041.6, B.08.041.7 and B.08.041.8 | ||
| (3) Cider; Wine; Honey Wine | (3) 500 p.p.m. | ||
| (4) 1,000 p.p.m. |
PART IV
Food Additives That May Be Used as Class IV Preservatives
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Ascorbic Acid | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| A.2 | Ascorbyl Palmitate | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) 0.02% of the fat content. If ascorbyl stearate is also used the total must not exceed 0.02% of the fat content | ||
| (4) Infant formula | (4) 0.001% as consumed | ||
| A.3 | Ascorbyl Stearate | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Margarine | (2) 0.02% of the fat content. If ascorbyl palmitate is also used the total must not exceed 0.02% of the fat content | ||
| B.1 | Butylated Hydroxyanisole (a mixture of 2-tertiarybutyl-4- hydroxyanisole and 3-tertiarybutyl-4- hydroxyanisole) | (1) Fats and oils, lard, shortening | (1) 0.02%. If butylated hydroxytoluene, propyl gallate or tertiary butyl hydroquinone, singly or in combination, is also used, the total must not exceed 0.02% |
| (2) Dried breakfast cereals; Dehydrated potato products | (2) 0.005%. If butylated hydroxytoluene or propyl gallate or both are also used, the total must not exceed 0.005% | ||
| (3) Chewing gum | (3) 0.02%. If butylated hydroxytoluene or propyl gallate or both are also used, the total must not exceed 0.02% | ||
| (4) Essential oils; Citrus oil flavours; Dry flavours | (4) 0.125%. If butylated hydroxytoluene or propyl gallate or both are also used, the total must not exceed 0.125% | ||
| (5) Citrus oils | (5) 0.5%. If butylated hydroxytoluene or propyl gallate or both are also used, the total must not exceed 0.5% | ||
| (6) Partially defatted pork fatty tissue; Partially defatted beef fatty tissue | (6) 0.0065%. If butylated hydroxytoluene is also used the total must not exceed 0.0065% | ||
| (7) Vitamin A liquids for addition to food | (7) 5 mg/1,000,000 International Units | ||
| (8) Dry beverage mixes; Dry dessert and confectionery mixes | (8) 0.009% | ||
| (9) Active dry yeast | (9) 0.1% | ||
| (10) 0.02% of the fat or the oil content of the food. If butylated hydroxytoluene or propyl gallate or both are also used, the total must not exceed 0.02% of the fat or the oil content of the food | ||
| (11) Dry Vitamin D preparations for addition to food | (11) 10 mg/1,000,000 International Units | ||
| (12) Margarine | (12) 0.01% of the fat content. If butylated hydroxytoluene or propyl gallate or both are also used the total must not exceed 0.01% of the fat content | ||
| (13) Dried cooked poultry meat | (13) 0.015% of the fat content. If propyl gallate or citric acid or both are also used, the total must not exceed 0.015% of the fat content. | ||
| B.2 | Butylated Hydroxytoluene (3,5-ditertiarybutyl-4-hydroxytoluene) | (1) Fats and oils, lard, shortening | (1) 0.02%. If butylated hydroxyanisole, propyl gallate or tertiary butyl hydroquinone, singly or in combination, is also used, the total must not exceed 0.02% |
| (2) Dried breakfast cereals; Dehydrated potato products | (2) 0.005%. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.005% | ||
| (3) Chewing gum | (3) 0.02%. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.02% | ||
| (4) Essential oils; Citrus oil flavours; Dry flavours | (4) 0.125%. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.125% | ||
| (5) Citrus oils | (5) 0.5%. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.5% | ||
| (6) Partially defatted pork fatty tissue; Partially defatted beef fatty tissue | (6) 0.0065%. If butylated hydroxyanisole is also used the total must not exceed 0.0065% | ||
| (7) Vitamin A liquids for addition to food | (7) 5 mg/1,000,000 International Units | ||
| (8) Parboiled rice | (8) 0.0035% | ||
| (9) 0.02% of the fat or the oil content of the food. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.02% of the fat or the oil content of the food | ||
| (10) Dry Vitamin D preparations for addition to food | (10) 10 mg/1,000,000 Internatinal Units | ||
| (11) Margarine | (11) 0.01% of the fat content. If butylated hydroxyanisole or propyl gallate or both are also used the total must not exceed 0.01% of the fat content | ||
| C.1 | Citric Acid | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Dried cooked poultry meat | (3) 0.015% of the fat content. If butylated hydroxyanisole or propyl gallate or both are also used, the total must not exceed 0.015% of the fat content. | ||
| C.1.01 | Citric Acid Esters of Mono- and Di-glycerides | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) 0.01% of the fat content. If monoglyceride citrate, monoisopropyl citrate or stearyl citrate, singly or in combination, is also used, the total must not exceed 0.01% of the fat content | ||
| C.1.1 | L-Cysteine | Nutritional supplements set out in section B.24.201 | Good Manufacturing Practice |
| C.2 | L-Cysteine Hydrochloride | Sulphite replacement formulations for prepared fruits and vegetables | Good Manufacturing Practice |
| G.1 | Gum Guaiacum | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| L.1 | Lecithin | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| L.2 | Lecithin Citrate | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| M.1 | Monoglyceride Citrate | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) 0.01% of the fat content. If citric acid esters of mono- and di-glycerides, monoisopropyl citrate or stearyl citrate, singly or in combination, is also used, the total must not exceed 0.01% of the fat content | ||
| M.2 | Monoisopropyl Citrate | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Margarine | (3) 0.01% of the fat content. If citric acid esters of mono- and di-glycerides, monoglyceride citrate or stearyl citrate, singly or in combination, is also used, the total must not exceed 0.01% of the fat content | ||
| P.1 | Propyl Gallate | (1) Fats and oils, lard, shortening | (1) 0.02%. If butylated hydroxyanisole, butylated hydroxytoluene or tertiary butyl hydroquinone, singly or in combination, is also used, the total must not exceed 0.02% |
| (2) Dried breakfast cereals; Dehydrated potato products | (2) 0.005%. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used, the total must not exceed 0.005% | ||
| (3) Chewing gum | (3) 0.02%. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used, the total must not exceed 0.02% | ||
| (4) Essential oils; Dry flavours | (4) 0.125%. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used, the total must not exceed 0.125% | ||
| (5) Citrus oils | (5) 0.5%. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used, the total must not exceed 0.5% | ||
| (6) 0.02% of the fat or the oil content of the food. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used, the total must not exceed 0.02% of the fat or the oil content of the food | ||
| (7) Margarine | (7) 0.01% of the fat content. If butylated hydroxyanisole or butylated hydroxytoluene or both are also used the total must not exceed 0.01% of the fat content | ||
| (8) Dried cooked poultry meat | (8) 0.015% of the fat content. If butylated hydroxyanisole or citric acid or both are also used the total must not exceed 0.015% of the fat content. | ||
| T.1 | Tartaric Acid | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| T.1A | Tertiary Butyl Hydroquinone | Fats and oils, lard, shortening | 0.02%. If butylated hydroxyanisole, butylated hydroxytoluene or propyl gallate, singly or in combination, is also used, the total must not exceed 0.02% |
| T.2 | Tocopherols (alpha-tocopherol; tocopherols concentrate, mixed) | (1) Fats and oils; Lard; Monoglycerides and diglycerides; Shortening | (1) Good Manufacturing Practice |
| (2) Good Manufacturing Practice | ||
| (3) Infant formula | (3) 0.001% as consumed |
- SOR/79-285, ss. 1 to 4
- SOR/79-660, s. 18
- SOR/79-752, s. 10
- SOR/80-500, s. 7
- SOR/81-565, s. 6
- SOR/81-934, ss. 13 to 15
- SOR/82-383, s. 11
- SOR/86-89, s. 7
- SOR/86-1020, s. 1
- SOR/87-138, ss. 1, 2
- SOR/87-469, s. 2
- SOR/89-198, ss. 12 to 16
- SOR/91-124, ss. 10 to 12
- SOR/92-226, s. 1
- SOR/92-591, s. 2(F)
- SOR/94-689, s. 2(F)
- SOR/95-592, s. 1
- SOR/96-241, s. 2
- SOR/97-148, s. 7
- SOR/97-191, s. 4
- SOR/98-459, s. 1
- SOR/99-289, ss. 1 to 4
- SOR/2003-156, s. 1
- SOR/2005-316, ss. 4 to 6
- SOR/2010-94, s. 8(E)
- SOR/2010-141, ss. 1, 2
- SOR/2010-264, s. 4
- SOR/2011-235, ss. 7 to 13, 16(F)
- SOR/2011-280, ss. 7, 8
- SOR/2012-43, ss. 37 to 42
- SOR/2012-104, ss. 16(F), 17, 18
TABLE XII
Food Additives That May Be Used as Sequestering Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Ammonium Citrate, dibasic | Unstandardized foods | Good Manufacturing Practice |
| A.2 | Ammonium Citrate, monobasic | Unstandardized foods | Good Manufacturing Practice |
| C.1 | Calcium Citrate | Unstandardized foods | Good Manufacturing Practice |
| C.2 | Calcium Disodium Ethylenediaminetetraacetate (Calcium Disodium EDTA) | (1) Ale; Beer; Malt liquor; Porter; Stout | (1) 25 p.p.m. calculated as the anhydrous form |
| (2) French dressing; Mayonnaise; Salad dressing; Unstandardized dressings; Unstandardized sauces | (2) 75 p.p.m. calculated as the anhydrous form | ||
| (3) Potato salad; Unstandardized sandwich spreads | (3) 100 p.p.m. calculated as the anhydrous form | ||
| (4) Canned shrimp; Canned tuna | (4) 250 p.p.m. calculated as the anhydrous form | ||
| (5) Canned crabmeat; Canned lobster; Canned salmon | (5) 275 p.p.m. calculated as the anhydrous form | ||
| (6) Margarine | (6) 75 p.p.m. calculated as the anhydrous form | ||
| (7) Canned clams | (7) 340 p.p.m. calculated as the anhydrous form | ||
| (8) Beans; Beans with pork; Canned legumes except canned green beans, canned peas and canned wax beans | (8) 365 p.p.m. calculated as the anhydrous form | ||
| (9) Canned sea snails; Canned snails | (9) 300 p.p.m. calculated as the anhydrous form | ||
| (10) Ready-to-drink teas; Soft drinks | (10) 33 p.p.m. calculated as the anhydrous form | ||
| (11) Pasteurized sous-vide potatoes | (11) 100 p.p.m., singly or in combination with disodium EDTA, calculated as the anhydrous disodium EDTA form | ||
| C.3 | [Repealed, SOR/2012-43, s. 43] | ||
| C.4 | Calcium Phosphate, monobasic | (1) Ice cream mix; Ice milk mix; Sherbet | (1) Good Manufacturing Practice |
| (2) Unstandardized dairy products | (2) Good Manufacturing Practice | ||
| C.5 | Calcium Phosphate, tribasic | Ice cream mix; Ice milk mix | Good Manufacturing Practice |
| C.6 | Calcium Phytate | Glazed fruit | Good Manufacturing Practice |
| C.7 | Citric Acid | (1) Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Frozen fish fillets; frozen minced fish; frozen comminuted fish | (3) 0.1% | ||
| D.1 | Disodium Ethylenediaminetetraacetate (Disodium EDTA) | (1) Dressing and sauces | (1) 70 p.p.m. |
| (2) Unstandardized sandwich spreads | (2) 90 p.p.m | ||
| (3) Beans; Beans with pork; Canned legumes except canned green beans, canned peas and canned wax beans | (3) 165 p.p.m. | ||
| (4) Dried banana products | (4) 265 p.p.m | ||
| (5) Aqueous suspensions of colour lake preparations for use in coating confectionery tablets | (5) 1% of the colour preparation | ||
| (6) Pasteurized sous-vide potatoes | (6) 100 p.p.m., singly or in combination with calcium disodium EDTA, calculated as anhydrous disodium EDTA | ||
| (7) Edible coating for sausages | (7) 80 p.p.m. in the edible coating, calculated on the basis of the whole sausage | ||
| D.2 | [Repealed, SOR/2012-43, s. 45] | ||
| G.1 | Glycine | Mono- and diglycerides | 0.02% |
| P.1 | Phosphoric Acid | Mono- and diglycerides | 0.02% |
| P.2 | Potassium Phosphate, monobasic | (1) Ice cream mix; Ice milk mix; Sherbet | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| (3) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (3) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| P.3 | Potassium Pyrophosphate, tetrabasic | (1) Meat tenderizers | (1) Good Manufacturing Practice |
| (2) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (2) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| P.4 | Potassium Phosphate, dibasic | Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | 0.5% total added phosphate, calculated as sodium phosphate, dibasic |
| S.1 | Sodium Acid Pyrophosphate | (1) Canned seafoods | (1) Used singly or in combination with sodium hexametaphosphate or sodium tripolyphosphate, or both, total added phosphate not to exceed 0.5% calculated as sodium phosphate, dibasic |
| (2) Ice cream mix; Ice milk mix | (2) Good Manufacturing Practice | ||
| (3) Injection or cover solution for the curing of poultry or poultry meat | (3) Good Manufacturing Practice, and in accordance with B.22.021(e) | ||
| (4) Pumping pickle for the curing of pork, beef and lamb cuts | (4) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (6) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| S.2 | Sodium Citrate | (1) Ice cream mix; Ice milk mix; Pumping pickle, cover pickle and dry cure employed in the curing of preserved meat or preserved meat by-product; Sherbet | (1) Good Manufacturing Practice |
| (2) Unstandardized foods | (2) Good Manufacturing Practice | ||
| S.3 | Sodium Hexametaphosphate | (1) Canned seafoods | (1) Used singly or in combination with sodium acid pyrophosphate or sodium tripolyphosphate, or both, total added phosphate not to exceed 0.5% calculated as sodium phosphate, dibasic |
| (2) Ice cream mix; Ice milk mix | (2) Good Manufacturing Practice | ||
| (3) Injection or cover solution for the curing of poultry or poultry meat | (3) Good Manufacturing Practice, and in accordance with B.22.021(e) | ||
| (4) Pumping pickle for the curing of pork, beef and lamb cuts | (4) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (6) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| (7) Liquid whey destined for the manufacture of concentrated or dried whey products | (7) 800 p.p.m. in the concentrated or dried whey products | ||
| S.4 | Sodium Phosphate, dibasic | (1) Ice cream mix; Ice milk mix; Sherbet | (1) Good Manufacturing Practice |
| (2) Injection or cover solution for the curing of poultry or poultry meat | (2) Good Manufacturing Practice, and in accordance with B.22.021(e) | ||
| (3) Pumping pickle for the curing of pork, beef and lamb cuts | (3) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (5) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| S.5 | Sodium Phosphate, monobasic | (1) Ice cream mix; Ice milk mix; Sherbet | (1) Good Manufacturing Practice |
| (2) Injection or cover solution for the curing of poultry or poultry meat | (2) Good Manufacturing Practice, and in accordance with B.22.021(e) | ||
| (3) Pumping pickle for the curing of pork, beef and lamb cuts | (3) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (5) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| S.6 | Sodium Pyrophosphate, tetrabasic | (1) Ice cream mix; Ice milk mix; Sherbet | (1) Good Manufacturing Practice |
| (2) Meat tenderizers | (2) Good Manufacturing Practice | ||
| (3) Injection or cover solution for the curing of poultry or poultry meat | (3) Good Manufacturing Practice, and in accordance with B.22.021(e) | ||
| (4) Pumping pickle for the curing of pork, beef and lamb cuts | (4) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (5) Unstandardized foods | (5) Good Manufacturing Practice | ||
| (6) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (6) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| S.7 | Sodium Tripolyphosphate | (1) Injection or cover solution for the curing of poultry or poultry meat | (1) Good Manufacturing Practice, and in accordance with B.22.021(e) |
| (2) Meat tenderizers | (2) Good Manufacturing Practice | ||
| (3) Pumping pickle for the curing of pork, beef and lamb cuts | (3) Good Manufacturing Practice, and in accordance with B.14.009(f) and B.14.031(h) | ||
| (4) Unstandardized foods | (4) Good Manufacturing Practice | ||
| (5) Solid cut meat; prepared meat; prepared meat by-product; solid cut poultry meat; prepared poultry meat; prepared poultry meat by-product | (5) 0.5% total added phosphate, calculated as sodium phosphate, dibasic | ||
| (6) Canned seafoods | (6) Used singly or in combination with sodium acid pyrophosphate or sodium hexametaphosphate, or both, total added phosphate not to exceed 0.5% calculated as sodium phosphate, dibasic | ||
| S.8 | Stearyl Citrate | Margarine | 0.01% of the fat content. If citric acid esters of mono- and di-glycerides, monoglyceride citrate or monoisopropyl citrate, singly or in combination, is also used, the total must not exceed 0.01% of the fat content |
- SOR/79-660, ss. 19, 20
- SOR/80-501, s. 4
- SOR/82-596, ss. 4 to 9
- SOR/94-141, s. 1
- SOR/94-262, ss. 4 to 12
- SOR/94-689, s. 2
- SOR/95-435, s. 2
- SOR/97-30, s. 1
- SOR/97-562, s. 1
- SOR/580, s. 1(F)
- SOR/2005-316, ss. 7 to 11
- SOR/2010-40, s. 2
- SOR/2010-142, ss. 57, 58
- SOR/2010-143, ss. 37(F), 38
- SOR/2011-235, s. 14
- SOR/2012-43, ss. 43 to 45
- SOR/2012-104, s. 19(F)
- SOR/2012-112, s. 1
TABLE XIII
Food Additives That May Be Used as Starch Modifying Agents
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Acetic Anhydride | Starch | Good Manufacturing Practice |
| A.2 | Adipic Acid | Starch | Good Manufacturing Practice |
| A.3 | Aluminum Sulphate | Starch | Good Manufacturing Practice |
| E.1 | [Repealed, SOR/2016-143, s. 2] | ||
| H.1 | Hydrochloric Acid | Starch | Good Manufacturing Practice |
| H.2 | Hydrogen Peroxide | Starch | Good Manufacturing Practice |
| M.1 | Magnesium Sulphate | Starch | 0.4% |
| N.1 | Nitric Acid | Starch | Good Manufacturing Practice |
| O.1 | Octenyl Succinic Anhydride (OSA) | Starch | Good Manufacturing Practice |
| P.1 | Peracetic Acid | Starch | Good Manufacturing Practice |
| P.2 | Phosphorus Oxychloride | Starch | Good Manufacturing Practice |
| P.3 | Potassium Permanganate | Starch | 50 p.p.m. of Manganese Sulphate calculated as Manganese |
| P.4 | Propylene Oxide | Starch | 25% |
| S.1 | Sodium Acetate | Starch | Good Manufacturing Practice |
| S.2 | Sodium Bicarbonate | Starch | Good Manufacturing Practice |
| S.3 | Sodium Carbonate | Starch | Good Manufacturing Practice |
| S.4 | Sodium Chlorite | Starch | Good Manufacturing Practice |
| S.5 | Sodium Hydroxide | Starch | Good Manufacturing Practice |
| S.6 | Sodium Hypochlorite | Starch | Good Manufacturing Practice |
| S.7 | Sodium Trimetaphosphate | Starch | 400 p.p.m. calculated as Phosphorus |
| S.7A | Sodium Tripolyphosphate | Starch | Total residual phosphate not to exceed 0.4% (calculated as Phosphorus) |
| S.8 | Succinic Anhydride | Starch | Good Manufacturing Practice |
| S.9 | Sulphuric Acid | Starch | Good Manufacturing Practice |
- SOR/94-689, s. 2(F)
- SOR/2012-106, s. 2
- SOR/2016-143, s. 2
TABLE XIV
Food Additives That May Be Used as Yeast Foods
| Item No. | Column I | Column II | Column III |
|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Level of Use | |
| A.1 | Ammonium Chloride | (1) Flour; Whole wheat flour | (1) 2,000 p.p.m. of the flour |
| (2) Bread | (2) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) | ||
| (3) Unstandardized foods | (3) Good Manufacturing Practice | ||
| A.2 | Ammonium Phosphate, dibasic | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Cider; Honey wine; Wine | (2) Good Manufacturing Practice | ||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | ||
| A.3 | Ammonium Phosphate, monobasic | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Ale; Beer; Cider; Honey wine; Light beer; Malt liquor; Porter; Stout; Wine | (2) Good Manufacturing Practice | ||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | ||
| A.4 | Ammonium Sulphate | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Cider; Honey wine; Wine | (2) Good Manufacturing Practice | ||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | ||
| C.1 | Calcium Carbonate | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| C.2 | Calcium Chloride | Unstandardized bakery products | Good Manufacturing Practice |
| C.3 | Calcium Citrate | Unstandardized bakery products | Good Manufacturing Practice |
| C.4 | Calcium Lactate | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| C.5 | Calcium Phosphate, dibasic | (1) Bread | (1) 2,500 p.p.m. of the flour. For combinations see paragraph B.13.021(m) |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| C.6 | Calcium Phosphate, monobasic | (1) Bread | (1) 7,500 p.p.m. of flour. For combinations see paragraph B.13.021(m) |
| (2) Flour | (2) 7,500 p.p.m. of flour | ||
| (3) Unstandardized bakery products | (3) Good Manufacturing Practice | ||
| C.7 | Calcium Phosphate, tribasic | Unstandardized bakery products | Good Manufacturing Practice |
| C.8 | Calcium Sulphate | (1) Bread | (1) 5,000 p.p.m. of the flour |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| F.1 | Ferrous Sulphate | Bacterial cultures | Good Manufacturing Practice |
| M.1 | Manganese Sulphate | Ale; Beer; Light beer; Malt liquor; Porter; Stout | Good Manufacturing Practice |
| P.1 | Phosphoric Acid | Ale; Beer; Light beer; Malt liquor; Porter; Stout | Good Manufacturing Practice |
| P.2 | Potassium Chloride | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| P.4 | Potassium Phosphate, dibasic | (1) Ale; Beer; Cider; Honey wine; Light beer; Malt liquor; Porter; Stout; Wine | (1) Good Manufacturing Practice |
| (2) Unstandardized bakery products | (2) Good Manufacturing Practice | ||
| P.5 | Potassium Phosphate, monobasic | Ale; Beer; Cider; Honey wine; Light beer; Malt liquor; Porter; Stout; Wine | Good Manufacturing Practice |
| S.1 | Sodium Sulphate | Unstandardized bakery products | Good Manufacturing Practice |
| U.1 | [Repealed, SOR/87-5, s. 1] | ||
| Z.1 | Zinc Sulphate | (1) Ale; Beer; Light beer; Malt liquor; Porter; Stout | (1) Good Manufacturing Practice |
| (2) Bacterial cultures | (2) Good Manufacturing Practice | ||
- SOR/87-5, s. 1
- SOR/94-689, s. 2(F)
- SOR/95-281, ss. 6, 7
- SOR/2010-41, ss. 7(E), 9(E)
TABLE XV
Food Additives That May Be Used as Carrier or Extraction Solvents
| Item No. | Column I | Column II | Column III | Column IV |
|---|---|---|---|---|
| Additive | Permitted in or Upon | Maximum Residue | Maximum Level of Use | |
| 1 | Acetone | (1) Spice extracts; Natural extractives | (1) 30 p.p.m. | |
| (2) Meat and Egg Marking Inks | (2) Good Manufacturing Practice | |||
| 2 | Benzyl Alcohol | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 3 | 1,3-Butylene Glycol | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 3.1 | Carbon Dioxide | (1) Green coffee beans and tea leaves for decaffeination purposes | (1) Good Manufacturing Practice | |
| (2) Spice extracts; Natural extractives; (naming the flavour) Flavour (Division 10); Hop extract in accordance with subparagraph B.02.130(b)(v) and paragraph B.02.133(b); Pre-isomerized hop extract in accordance with subparagraph B.02.134(1)(a)(ii) | (2) Good Manufacturing Practice | |||
| (3) Egg Products | (3) Good Manufacturing Practice | |||
| (4) Cocoa powder | (4) Good Manufacturing Practice | |||
| 4 | Castor Oil | Oil-soluble annatto; Annatto butter colour; Annatto margarine colour | Good Manufacturing Practice | |
| 4.1 | Citric Acid Esters of Mono- and Di-glycerides | (1) Natural extractives; Spice extracts | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 5 | Ethyl Acetate | (1) Spice extracts; Natural extractives; (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| (3) Green coffee beans for decaffeination purposes | (3) 10 p.p.m. in both roasted and decaffeinated soluble (instant) coffee | |||
| (4) Tea leaves for decaffeination purposes | (4) 50 p.p.m. | |||
| 6 | Ethyl Alcohol (Ethanol) | (1) Spice extracts; Natural extractives; (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| (3) Colour mixtures and preparations (Division 6) | (3) Good Manufacturing Practice | |||
| (4) Meat and Egg Marking Inks | (4) Good Manufacturing Practice | |||
| (5) Food additive preparations | (5) Good Manufacturing Practice | |||
| (6) Hop extract in accordance with subparagraph B.02.130(b)(v) and paragraph B.02.133(b); Pre-isomerized hop extract in accordance with subparagraph B.02.134(1)(a)(iii) | (6) Good Manufacturing Practice | |||
| 6.A | Ethyl alcohol denatured with methanol | Vegetable oil seed meals | 10 p.p.m. methanol | |
| 7 | [Repealed, SOR/82-406, s. 1] | |||
| 8 | Glycerol (Glycerin) | (1) (naming the flavour) Extract; (naming the flavour) Essence; (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| (3) Colour mixtures and preparations (Division 6) | (3) Good Manufacturing Practice | |||
| (4) Food additive preparations | (4) Good Manufacturing Practice | |||
| 9 | Glyceryl diacetate | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 10 | Glyceryl triacetate (Triacetin) | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 11 | Glyceryl tributyrate (Tributyrin) | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 12 | Hexane | (1) Spice extracts; Natural extractives | (1) 25 p.p.m. | |
| (2) Hop extract in accordance with subparagraph B.02.130(b)(v) and paragraph B.02.133(a) | (2) 2.2% | |||
| (3) Vegetable fats and oils | (3) 10 p.p.m. | |||
| (4) Vegetable oil seed meals | (4) 10 p.p.m. | |||
| (5) Pre-isomerized hop extract in accordance with subparagraph B.02.134(1)(a)(i) and subsection B.02.134(2) | (5) 1.5 p.p.m. per percent iso-alpha acid content of the pre-isomerized hop extract | |||
| 13 | Isopropyl alcohol (Isopropanol) | (1) Spice extracts; Natural extractives | (1) 50 p.p.m. | |
| (2) Fish protein | (2) 0.15% | |||
| (3) (naming the flavour) Flavour (Division 10) | (3) Good Manufacturing Practice | |||
| (4) Unstandardized flavouring preparations | (4) Good Manufacturing Practice | |||
| (5) Meat and Egg Marking Inks | (5) Good Manufacturing Practice | |||
| 14 | Methyl Alcohol (methanol) | (1) Spice extracts; Natural extractives | (1) 50 p.p.m. | |
| (2) Hop extract in accordance with subparagraph B.02.130(b)(v) and paragraph B.02.133(a) | (2) 2.2% | |||
| (3) Meat and Egg Marking Inks | (3) Good Manufacturing Practice | |||
| 14.1 | Methyl ethyl ketone (2-Butanone) | Spice extracts; Natural extractives | 50 p.p.m. | |
| 15 | Methylene Chloride (Dichloro-methane) | (1) Spice extracts; Natural extractives | (1) 30 p.p.m. | |
| (2) Hop extract in accordance with subparagraph B.02.130(b)(v) and paragraph B.02.133(a) | (2) 2.2% in hop extract | |||
| (3) Green coffee beans and Tea leaves for decaffeination purposes | (3) 10 p.p.m. in decaffeinated roasted coffee, decaffeinated soluble (instant) coffee, decaffeinated tea leaves and decaffeinated instant tea | |||
| 16 | Monoglycerides and diglycerides | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Oil-soluble annatto; Annatto butter colour; Annatto margarine colour | (2) Good Manufacturing Practice | |||
| (3) Unstandardized flavouring preparations | (3) Good Manufacturing Practice | |||
| (4) Food additive preparations | (4) Good Manufacturing Practice | |||
| 17 | Monoglyceride citrate | (1) Spice extracts; Natural extractives | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
| 18 | [Repealed, SOR/2016-143, s. 3] | |||
| 19 | 1,2-Propylene glycol (1,2-propanediol) | (1) (naming the flavour) Extract; (naming the flavour) Essence; (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Oil-soluble annatto; Annatto butter colour; Annatto margarine colour | (2) Good Manufacturing Practice | |||
| (3) Unstandardized flavouring preparations | (3) Good Manufacturing Practice | |||
| (4) Colour mixtures and preparations (Division 6) | (4) Good Manufacturing Practice | |||
| (5) Food additive preparations | (5) Good Manufacturing Practice | |||
| 20 | Propylene glycol mono-esters and diesters of fat-forming fatty acids | Oil-soluble annatto; Annatto butter colour; Annatto margarine colour | Good Manufacturing Practice | |
| 21 | Triethyl-citrate | (1) (naming the flavour) Flavour (Division 10) | (1) Good Manufacturing Practice | |
| (2) Unstandardized flavouring preparations | (2) Good Manufacturing Practice | |||
- SOR/78-403, ss. 26, 27
- SOR/82-383, s. 12
- SOR/82-406, s. 1
- SOR/82-913, s. 5
- SOR/82-1071, ss. 21, 22
- SOR/84-541, s. 1
- SOR/86-89, ss. 8, 9
- SOR/86-178, ss. 4 to 7
- SOR/86-1112, s. 9
- SOR/90-667, s. 1
- SOR/94-689, s. 2
- SOR/96-259, s. 1
- SOR/96-377, s. 1
- SOR/2011-235, s. 15
- SOR/2016-143, s. 3
DIVISION 17Salt
B.17.001 (1) [S]. Salt, other than crude rock salt, shall be crystalline sodium chloride and may contain
(a) one or more of the following anti-caking agents,
(i) calcium aluminum silicate, calcium phosphate tribasic, calcium silicate, calcium stearate, magnesium carbonate, magnesium silicate, magnesium stearate, silicon dioxide and sodium aluminum silicate, the total amount not to exceed one per cent and, in the case of fine grained salt, the total amount not to exceed two per cent,
(ii) propylene glycol in an amount not exceeding 0.035 per cent, and
(iii) sodium ferrocyanide decahydrate in an amount not exceeding 13 parts per million calculated as anhydrous sodium ferrocyanide;
(b) not more than
(i) 1.4 per cent, singly or in combination, of calcium sulphate or potassium chloride,
(ii) 13 parts per million anhydrous sodium ferrocyanide when added as sodium ferrocyanide decahydrate in the production of dendritic crystals of salt,
(iii) 10 parts per million of polyoxyethylene (20) sorbitan monooleate when used in the production of coarse crystal salt,
(iv) 15 parts per million of sodium alginate when used in the production of coarse crystal salt, and
(v) 0.1 per cent other ingredients; and
(c) notwithstanding paragraphs (a) and (b), the total level of sodium ferrocyanide decahydrate, whether added as an anti-caking agent or as an adjuvant in the production of dendritic salt, shall not exceed 13 parts per million, calculated as anhydrous sodium ferrocyanide.
(2) [Repealed, SOR/97-151, s. 26]
- SOR/79-662, s. 18
- SOR/86-1125, s. 3
- SOR/97-151, s. 26
B.17.002 [Repealed, SOR/79-662, s. 18]
B.17.003 Notwithstanding section B.17.001, salt for table or general household use shall contain 0.01 per cent potassium iodide, with or without dextrose, sodium thiosulphate or sodium bicarbonate as a stabilizer of the iodide and the presence of iodide shall be shown on the principal display panel.
DIVISION 18
Sweetening Agents
B.18.001 [S]. Sugar
(a) shall be the food chemically known as sucrose; and
(b) shall contain not less than 99.8 per cent sucrose.
B.18.002 [S]. Liquid Sugar shall be the food obtained by dissolving sugar in water.
B.18.003 [S]. Invert Sugar shall be the food obtained by the partial or complete hydrolysis of sugar.
B.18.004 [S]. Liquid Invert Sugar shall be the food consisting of a solution of invert sugar in water.
B.18.005 [S]. No person shall sell liquid sugar or liquid invert sugar unless the label carries a statement of the percentage of sugar or invert sugar contained therein.
B.18.006 [S]. Icing Sugar
(a) shall be powdered sugar; and
(b) may contain
(i) food colour, and
(ii) either not more than five per cent starch or an anticaking agent.
B.18.007 [S]. Brown Sugar, Yellow Sugar or Golden Sugar
(a) shall be the food obtained from the syrups originating in the sugar refining process;
(b) may contain not more than
(i) 4.5 per cent moisture, and
(ii) 3.5 per cent sulphate ash; and
(c) shall not contain less than 90 per cent sugar and invert sugar.
B.18.008 [S]. Refined Sugar Syrup, Refiners’ Syrup or Golden Syrup
(a) shall be the food made from syrup originating in the sugar refining process;
(b) may be hydrolyzed; and
(c) shall not contain more than
(i) 35 per cent moisture, and
(ii) 2.5 per cent sulphated ash.
B.18.009 [S]. Fancy Molasses
(a) shall be the syrupy food obtained by the evaporation and partial inversion of the clarified or unclarified sugar cane juice from which sugar has not been previously extracted;
(b) may contain sulphurous acid or its salts; and
(c) shall not contain more than
(i) 25 per cent moisture, and
(ii) 3 per cent sulphated ash.
B.18.010 [S]. Table Molasses
(a) shall be the liquid food obtained in the process of manufacturing raw or refined sugar;
(b) may contain sulphurous acid or its salts;
(c) shall not contain more than
(i) 25 per cent moisture, and
(ii) 3 per cent sulphated ash.
B.18.011 [S]. Refiners’ Molasses, Blackstrap Molasses or Cooking Molasses
(a) shall be the residual liquid food obtained in the process of manufacturing raw or refined sugar;
(b) may contain sulphurous acid or its salts;
(c) shall not contain more than
(i) 25 per cent moisture, and
(ii) 12 per cent sulphated ash.
B.18.015 [S]. (1) Dextrose Anhydrous, for the purpose of Part B of these Regulations
(a) shall be the food chemically known as dextrose;
(b) shall contain not less than 99.5 per cent D-glucose on a dry basis;
(c) shall contain not more than 0.25 per cent sulphated ash on a dry basis;
(d) shall contain not less than 98 per cent total solids; and
(e) may contain sulphurous acid or its salts.
(2) Dextrose Monohydrate, for the purpose of Part B of these Regulations
(a) shall be the food chemically known as dextrose;
(b) shall contain not less than 99.5 per cent D-glucose on a dry basis;
(c) shall contain not more than 0.25 per cent sulphated ash on a dry basis;
(d) shall contain not less than 90 per cent total solids; and
(e) may contain sulphurous acid or its salts.
- SOR/84-300, s. 51
B.18.016 [S]. Glucose or Glucose Syrup
(a) shall be the purified concentrated solution of nutritive saccharides obtained from the incomplete hydrolysis, by means of acid or enzymes, of starch or of a starch-containing substance;
(b) shall have a total solids content of not less than 70 per cent;
(c) shall have a sulphated ash content of not more than 1.0 per cent on a dry basis;
(d) shall have a reducing sugar content (dextrose equivalent) of not less than 20 per cent expressed as D-glucose on a dry basis; and
(e) may contain sulphurous acid or its salts.
- SOR/78-402, s. 8
B.18.017 [S]. Glucose Solids or Dried Glucose Syrup
(a) shall be glucose or glucose syrup from which the water has been partially removed;
(b) shall have a total solids content of not less than 93 per cent;
(c) shall have a sulphated ash content of not more than 1.0 per cent on a dry basis;
(d) shall have a reducing sugar content (dextrose equivalent) of not less than 20 per cent expressed as D-glucose, on a dry basis; and
(e) may contain sulphurous acid or its salts.
B.18.018 [S]. (Naming the source of the glucose) Syrup
(a) shall be glucose;
(b) may contain
(i) a sweetening agent,
(ii) a flavouring preparation,
(iii) sorbic acid,
(iv) sulphurous acid or its salts,
(v) salt, and
(vi) water; and
(c) shall not contain more than
(i) 35 per cent moisture; and
(ii) 3 per cent ash.
- SOR/94-83, s. 1
B.18.019 [S]. Lactose
(a) shall be the carbohydrate normally obtained from whey and may
(i) be anhydrous,
(ii) contain one molecule of water of crystalization, or
(iii) be a mixture of both subparagraphs (i) and (ii);
(b) shall not contain less than 99.0 per cent anhydrous lactose on a moisture free basis;
(c) shall not contain more than 0.3 per cent sulphated ash on a moisture free basis;
(d) shall have a weight loss of not more than 6.0 per cent on drying; and
(e) shall have, in a 10 per cent solution, a pH of not less than 4.5 and not more than 7.0.
Honey
B.18.025 [S]. Honey shall be the food produced by honey bees and derived from
(a) the nectar of blossoms,
(b) secretions of living plants, or
(c) secretions on living plants,
and shall have
(d) a fluid, viscous or partly or wholly crystallized consistency;
(e) a diastase activity, determined after processing and blending, as represented by a diastase figure on the Gothe scale of not less than eight where the hydroxy-methyl-furfural content is not more than 0.004 per cent; or
(f) a diastase activity, determined after processing and blending, as represented by a diastase figure on the Gothe scale of not less than three where the hydroxy-methyl-furfural content is not more than 0.0015 per cent.
B.18.026 (1) Subject to subsection (2), honey derived mainly from nectar of blossoms shall not contain
(a) less than 65 per cent apparent reducing sugar, calculated as invert sugar;
(b) more than 20 per cent moisture;
(c) more than 5 per cent apparent sucrose;
(d) more than 0.1 per cent water insoluble solids, except that pressed honey shall contain not more than 0.5 per cent water insoluble solids;
(e) more than 0.6 per cent ash; and
(f) more than 40 milliequivalents acid per 1 000 grams.
(2) Honey derived mainly from the nectar of lavender, rubinia, alfalfa, or banksia menziesii shall meet the requirements of paragraphs (1)(a), (b) and (d) to (f) and shall contain not more than 10 per cent apparent sucrose.
B.18.027 Honey derived from secretions of living plants or from secretions on living plants shall not contain
(a) less than 60 per cent apparent reducing sugar, calculated as invert sugar;
(b) more than 20 per cent moisture;
(c) more than 10 per cent apparent sucrose;
(d) more than 0.1 per cent water insoluble solids, except that pressed honey shall contain not more than 0.5 per cent water insoluble solids;
(e) more than 1.0 per cent ash; and
(f) more than 40 milliequivalents acid per 1 000 grams.
- SOR/84-300, s. 52
DIVISION 19Vinegar
B.19.001 Vinegar shall be the liquid obtained by the acetous fermentation of an alcoholic liquid and shall contain not less than 4.1 per cent and not more than 12.3 per cent acetic acid.
- SOR/92-626, s. 16
- SOR/93-243, s. 2
B.19.002 The percentage of acetic acid by volume contained in any vinegar described in Division 19 shall be shown on the principal display panel followed by the words “acetic acid”.
B.19.003 [S]. Wine Vinegar shall be vinegar made from wine and may contain caramel.
B.19.004 [S]. Spirit Vinegar, Alcohol Vinegar, White Vinegar or Grain Vinegar shall be vinegar made from diluted distilled alcohol.
B.19.005 [S]. Malt Vinegar shall be vinegar made from an infusion of malt undistilled prior to acetous fermentation, may contain other cereals or caramel, shall be dextro-rotatory, and shall contain, in 100 millilitres measured at a temperature of 20°C, not less than
(a) 1.8 grams of solids, and
(b) 0.2 gram of ash.
B.19.006 [S]. Cider Vinegar or Apple Vinegar shall be vinegar made from the liquid expressed from whole apples, apple parts or apple culls and may contain caramel.
B.19.007 [S]. Blended Vinegar shall be a combination of two or more varieties of vinegar of which spirit vinegar shall contribute not more than 55 per cent of the total acetic acid.
B.19.008 No person shall name any of the varieties of vinegar forming a blended vinegar unless the label of such blended vinegar carries a complete list of all the varieties of vinegar present, in descending order of proportionate content, based on acetic acid.
B.19.009 The maximum limits for the acetic acid content of a vinegar described in section B.19.001 do not apply to vinegar sold only for manufacturing use if the words “For Manufacturing Use Only” are shown on the principal display panel and upon all documents pertaining to such vinegar.
DIVISION 20Tea
B.20.001 [S]. Tea shall be the dried leaves and buds of Thea sinensis (L.) Sims prepared by the usual trade processes.
B.20.002 [S]. Black Tea shall be black tea or a blend of two or more black teas and shall contain, on the dry basis, not less than 30 per cent water-soluble extractive, as determined by official method FO-37, Determination of Water-Soluble Extractive in Tea, October 15, 1981, and not less than four per cent and not more than seven per cent total ash.
- SOR/82-768, s. 61
B.20.003 The provisions of B.20.002 do not apply to original unblended black tea that contains, on the dry basis, not less than 25 per cent water-soluble extractive, as determined by official method FO-37, Determination of Water-Soluble Extractive in Tea, October 15, 1981, and not less than four per cent and not more than seven per cent total ash, and is packaged according to good commercial practice in the country of origin.
- SOR/82-768, s. 61
B.20.004 [S]. Green Tea shall contain, on the dry basis, not less than 33 per cent water-soluble extractive, as determined by official method FO-37, Determination of Water-Soluble Extractive in Tea, October 15, 1981, and not less than four per cent and not more than seven per cent total ash.
- SOR/82-768, s. 61
B.20.005 [S]. Decaffeinated (indicating the type of tea)
(a) shall be tea of the type indicated, from which caffeine has been removed and that, as a result of the removal, contains not more than 0.4 per cent caffeine; and
(b) may have been decaffeinated by means of extraction solvents set out in Table XV to Division 16.
- SOR/90-429, s. 2
DIVISION 21Marine and Fresh Water Animal Products
B.21.001 The foods referred to in this Division are included in the term marine and fresh water animal products.
B.21.002 In this Division,
- filler
filler means
(a) flour or meal prepared from grain or potato, but not from a legume,
(b) processed wheat flour containing not less than the equivalent of 80 per cent dextrose, as determined by official method FO-32, Determination of Fillers, Binders and Dextrose Equivalent, October 15, 1981,
(c) bread, biscuit or bakery products, but not those containing or made with a legume,
(d) milk powder, skim milk powder, buttermilk powder or whey powder, and
(e) starch; (remplissage)
- marine and fresh water animal
marine and fresh water animal includes
(a) fish,
(b) crustaceans, molluscs, other marine invertebrates,
(c) marine mammals, and
(d) frogs. (animaux marins et animaux d’eau douce)
- SOR/82-768, s. 62
B.21.003 [S]. Fish shall be the clean, dressed edible portion of fish, with or without salt or seasoning, and may
(a) in the case of frozen fillets, contain ascorbic acid or its sodium salt, citric acid, or erythorbic acid or its sodium salt, and
(i) sodium tripolyphosphate, sodium hexametaphosphate or a combination of sodium tripolyphosphate, sodium acid pyrophosphate and sodium pyrophosphate tetrabasic, or
(ii) a mixture of sodium hexametaphosphate and sodium carbonate;
(b) if frozen, have a glaze consisting of water, acetylated monoglycerides, calcium chloride, sodium alginate, sodium carboxymethyl cellulose, sodium phosphate (dibasic), corn syrup, dextrose, glucose, glucose solids, ascorbic acid or its sodium salt or erythorbic acid or its sodium salt; and
(c) if frozen minced, contain sodium tripolyphosphate, sodium hexametaphosphate, ascorbic acid or its sodium salt, citric acid, erythorbic acid or its sodium salt, or a combination of sodium tripolyphosphate, sodium acid pyrophosphate and sodium pyrophosphate tetrabasic.
- SOR/84-300, s. 53
- SOR/88-534, s. 7
- SOR/91-149, s. 4
- SOR/97-562, s. 2
- SOR/2000-353, s. 9(F)
- SOR/2017-18, s. 6(F)
B.21.004 [S]. In this Division, meat shall be the clean, dressed flesh of crustaceans, molluscs, other marine invertebrates and marine mammals, whether minced or not, with or without salt or seasoning, and in the case of frozen lobster, frozen crab, frozen shrimp and frozen clams, may contain sodium tripolyphosphate or sodium hexametaphosphate or a combination of sodium 0hexametaphosphate and sodium carbonate or a combination of sodium tripolyphosphate, sodium acid pyrophosphate and sodium pyrophosphate tetrabasic.
- SOR/88-534, s. 8
- SOR/91-149, s. 5
B.21.005 Fish, except fish protein, and meat products or preparations thereof are adulterated if any of the following substances or any substance in one of the following classes is present therein or has been added thereto:
(a) mucous membranes, any organ or portion of the genital system, or any organ or portion of a marine or fresh water animal that is not commonly sold as an article of food;
(b) preservatives, other than those provided for in this Division, except
(i) sorbic acid or its salts in dried fish that has been smoked or salted, and in cold processed smoked and salted fish paste, and
(ii) benzoic acid or its salts, methyl-p-hydroxy benzoate and propyl-p-hydroxy benzoate in marinated or similar cold-processed, packaged fish and meat products; and
(c) food colour except as provided for in this Division.
- SOR/2017-18, ss. 7(F), 20(F)
B.21.006 [S]. Prepared fish or prepared meat shall be the whole or minced food prepared from fresh or preserved fish or meat respectively, may be canned or cooked, and may,
(a) in the case of lobster paste and fish roe (caviar), contain food colour;
(b) in the case of canned shellfish, canned spring mackerel and frozen cooked shrimp, contain citric acid or lemon juice;
(c) in the case of fish paste, contain filler, fish binder, monoglycerides or mono and diglycerides;
(d) in the case of canned crabmeat, lobster, salmon, shrimp and tuna, contain aluminum sulphate and calcium disodium ethylenediaminetetraacetate;
(e) in the case of canned tuna, contain ascorbic acid;
(f) in the case of canned seafoods, contain sodium acid pyrophosphate, sodium hexametaphosphate or sodium tripolyphosphate, singly, or in combination, at a maximum level of total added phosphate not to exceed 0.5%, calculated as sodium phosphate, dibasic;
(g) contain liquid smoke flavour or liquid smoke flavour concentrate;
(h) contain edible oil, vegetable broth and tomato sauce or puree;
(i) contain a gelling agent if the principal display panel carries the word “jellied” as an integral part of the common name;
(j) contain salt;
(k) in the case of canned clams, canned sea snails and canned snails, contain calcium disodium ethylenediaminetetraacetate;
(l) in the case of canned flaked tuna, contain sodium sulphite;
(m) in the case of lumpfish caviar, contain tragacanth gum;
(n) in the case of a blend of prepared fish and prepared meat that has the appearance and taste of the flesh of a marine or freshwater animal, contain filler, fish binder, whole egg, egg-white, egg-yolk, food colour, gelling or stabilizing agents, texture-modifying agents, natural and artificial flavouring preparations, pH-adjusting agents, sweetener and, in a proportion not exceeding two per cent of the blend, a legume;
(o) in the case of crustaceans, contain potassium bisulphite, potassium metabisulphite, sodium bisulphite, sodium dithionite, sodium metabisulphite, sodium sulphite or sulphurous acid;
(p) in the case of frozen crustaceans and molluscs, contain calcium oxide and sodium hydroxide;
(q) in the case of frozen pre-cooked battered or breaded fish products, contain citric acid at a level of use not exceeding 0.1 per cent;
(r) in the case of canned clams, contain sodium erythorbate at a level of use not exceeding 350 parts per million;
(s) in the case of minced products, other than lumpfish caviar, contain tragacanth gum at a level of use not exceeding 0.75 per cent; and
(t) contain a Class II preservative.
- SOR/80-13, s. 9
- SOR/81-60, s. 12
- SOR/84-602, s. 4
- SOR/86-1020, s. 2
- SOR/89-197, s. 2
- SOR/92-344, s. 6
- SOR/93-276, s. 13
- SOR/94-141, s. 2
- SOR/94-567, s. 3
- SOR/94-689, s. 2(E)
- SOR/97-151, s. 27
- SOR/97-562, s. 3
- SOR/2005-316, s. 12
- SOR/2007-76, s. 4
- SOR/2011-280, s. 9
- SOR/2012-43, s. 46
- SOR/2017-18, ss. 8, 9(F), 17
- SOR/2018-69, s. 6
B.21.007 [S]. Fish binder for use in or upon prepared fish or prepared meat shall be filler with any combination of salt, sugar, dextrose, glucose, spices and other seasonings.
- SOR/2017-18, s. 10(F)
- SOR/2018-69, s. 7(F)
B.21.008 No person shall sell filler or a fish binder represented for use in fish products either by label or in any advertisement unless the label carries adequate directions for use in accordance with the limits provided in section B.21.020.
B.21.009 Powdered fully hydrogenated cottonseed oil may be applied as a release agent to the surface of marine and fresh water animal products in an amount not greater than 0.25% of the product.
- SOR/2010-142, s. 59(F)
- SOR/2022-168, s. 43
Prepared Fish
B.21.020 No person shall sell prepared fish or prepared meat that contains more than
(a) that amount of filler, fish binder or other ingredients that is represented by four per cent reducing sugars, calculated as dextrose, as determined by official method FO-32, Determination of Fillers, Binders and Dextrose Equivalent, October 15, 1981; and
(b) 70 per cent moisture where such prepared fish contains filler.
- SOR/82-768, s. 63
- SOR/2017-18, s. 20(F)
B.21.021 [S]. Preserved fish or preserved meat shall be cooked or uncooked fish or meat that is dried, salted, pickled, cured or smoked and may contain a Class I preservative, a Class II preservative, dextrose, glucose, spices, sugar and vinegar, and
(a) dried fish that has been smoked or salted, and cold-processed smoked and salted fish paste may contain sorbic acid or its salts;
(b) smoked fish may contain food colour;
(c) packaged fish and meat products that are marinated or otherwise cold-processed may contain saunderswood (sandalwood), benzoic acid or its salts, methyl-p-hydroxy benzoate and propyl-p-hydroxy benzoate;
(d) salted anchovy, salted scad and salted shrimp may contain erythrosine in such amount as will result in the finished product containing not more than 125 parts per million of erythrosine; and
(e) minced products may contain tragacanth gum at a level of use not exceeding 0.75 per cent.
- SOR/95-493, s. 2
- SOR/97-562, s. 4(F)
- SOR/2007-76, s. 5
- SOR/2011-280, s. 10
- SOR/2017-18, ss. 11(F), 17, 20(F)
- SOR/2018-69, s. 8(F)
B.21.022 and B.21.023 [Repealed, SOR/79-252, s. 4]
B.21.024 Notwithstanding section B.21.020 lobster paste shall not contain more than two per cent filler or fish binder.
B.21.025 No person shall sell marine and fresh water animals, or marine and fresh water animal products, that are packed in a container that has been sealed to exclude air and that are smoked or to which liquid smoke flavour or liquid smoke flavour concentrate has been added, unless
(a) the container has been heat-processed after sealing at a temperature and for a time sufficient to destroy all spores of the species Clostridium botulinum;
(b) the contents of the container contain not less than nine per cent salt, as determined by official method FO-38, Determination of Salt in Smoked Fish, dated March 15, 1985;
(c) the contents of the container are customarily cooked before eating; or
(d) the contents of the container are frozen and the principal display panel of the label of the container carries the statement “Keep Frozen Prior to Use” in the same size type used for the common name of the contents of the container.
- SOR/80-13, s. 10
- SOR/82-566, s. 5
- SOR/82-768, s. 64
- SOR/89-198, s. 17
- SOR/94-567, s. 4
B.21.027 [S]. Fish Protein
(a) shall be the food prepared by
(i) extracting water, fat and other soluble components through the use of isopropyl alcohol from fresh whole edible fish of the order Clupeiformes, families Clupeidae and Osmeridae and the order Gadiformes, family Gadidae, or from trimmings resulting from the filleting of such fish when eviscerated, and
(ii) drying and grinding the protein concentrate resulting from the operation described in subparagraph (i);
(b) may contain a pH adjusting agent; and
(c) shall not contain
(i) less than 75 per cent protein, which protein shall be at least equivalent to casein in protein quality, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981.
(ii) and (iii) [Repealed, SOR/97-148, s. 8]
- SOR/82-768, s. 65
- SOR/97-148, s. 8
Froglegs
B.21.031 No person shall sell fresh or frozen froglegs unless they are free from bacteria of the genus Salmonella, as determined by official method MFO-10, Microbiological Examination of Froglegs, November 30, 1981.
- SOR/82-768, s. 66
DIVISION 22Poultry, Poultry Meat, Their Preparations and Products
B.22.001 [S]. Poultry shall be any bird that is commonly used as food.
B.22.002 [S]. Poultry meat shall be the clean, dressed flesh including the heart and gizzard of eviscerated poultry that is healthy at the time of slaughter.
- SOR/80-13, s. 11
B.22.003 [S]. Poultry meat by-product shall be the clean parts of poultry other than poultry meat commonly used as food and includes liver and skin but excludes the oesophagus, feet and head.
- SOR/80-13, s. 12
B.22.004 [S]. Giblets shall be the heart, liver and gizzard of poultry.
B.22.005 Poultry meat, poultry meat by-products or preparations thereof are adultered if any of the following substances or any substance in the following classes is present therein or has been added thereto:
(a) any organ or portion of poultry that is not commonly sold as food;
(b) preservatives, other than those provided for in this Division; and
(c) colour, other than caramel.
B.22.006 [S]. Prepared poultry meat or a prepared poultry meat by-product shall be any poultry meat or any poultry meat by-product, respectively, whether comminuted or not, to which has been added any ingredient permitted by these Regulations or that has been preserved, placed in a hermetically-sealed container or cooked, and may contain
(a) where a minimum total protein content or minimum meat protein requirement is prescribed in this Division, phosphate salts that do not when calculated as sodium phosphate, dibasic, exceed the maximum level provided therefor in Table XII to section B.16.100 and that are one or more of the following phosphate salts, namely,
(i) sodium acid pyrophosphate,
(ii) sodium hexametaphosphate,
(iii) sodium phosphate, dibasic,
(iv) sodium phosphate, monobasic,
(v) sodium pyrophosphate, tetrabasic,
(vi) sodium tripolyphosphate,
(vii) potassium phosphate, monobasic,
(viii) potassium phosphate, dibasic, and
(ix) potassium pyrophosphate, tetrabasic;
(a.1) a Class II preservative;
(b) in the case of dried, cooked poultry meat, a Class IV preservative; and
(c) in the case of vacuum-packed sliced cooked turkey, Carnobacterium maltaromaticum CB1.
- SOR/81-934, s. 16
- SOR/94-262, s. 13
- SOR/2010-264, s. 5
- SOR/2011-280, s. 11
B.22.008 In this Division, filler means any vegetable material (except tomato or beetroot), milk, egg, yeast, or any derivative or combination thereof that is acceptable as food.
- SOR/82-768, s. 67
- SOR/84-300, s. 54(E)
- SOR/86-875, s. 6
B.22.009 No person shall sell
(a) any poultry intended for consumption as food if any preparation having oestrogenic activity has been administered to the poultry; or
(b) poultry meat or poultry meat by-product that contains any residues of exogenous oestrogenic substances.
- SOR/87-626, s. 2
B.22.010 Powdered fully hydrogenated cottonseed oil may be applied as a release agent to the surface of poultry meat, poultry meat by-product, prepared poultry meat, prepared poultry meat by-product, extended poultry product and simulated poultry product in an amount not greater than 0.25% of the product.
- SOR/2010-142, s. 59(F)
- SOR/2022-168, s. 44
B.22.011 [S]. Solid cut poultry meat shall be
(a) a whole cut of poultry meat; or
(b) a product consisting of pieces of poultry meat of which at least 80 per cent weigh at least 25 g each.
- SOR/94-262, s. 14
B.22.012 (1) No person shall sell solid cut poultry meat to which phosphate salts or water has been added unless
(a) that meat
(i) where cooked, contains a meat protein content of not less than 12 per cent, and
(ii) where uncooked, contains a meat protein content of not less than 10 per cent; and
(b) that meat contains, phosphate salts that do not when calculated as sodium phosphate, dibasic, exceed the maximum level provided therefor in Table XII to section B.16.100 and that are one or more of the following phosphate salts, namely,
(i) sodium acid pyrophosphate,
(ii) sodium hexametaphosphate,
(iii) sodium phosphate, dibasic,
(iv) sodium phosphate, monobasic,
(v) sodium pyrophosphate, tetrabasic,
(vi) sodium tripolyphosphate,
(vii) potassium phosphate, monobasic,
(viii) potassium phosphate, dibasic, and
(ix) potassium pyrophosphate, tetrabasic.
(2) A bone or a visible fat layer shall not be included in any calculation used to determine meat protein content for the purposes of paragraph (1)(a).
- SOR/94-262, s. 14
B.22.013 No person shall sell the whole or any part of a dressed poultry carcass that has been placed in a chilling tank containing fluids to which phosphate salts have been added.
- SOR/94-262, s. 14
Poultry Meat Stews
B.22.016 For the purposes of sections B.22.017 to B.22.019, stew poultry meat means cooked or uncooked poultry meat containing not more than 15 per cent fat, calculated on the weight of uncooked stew poultry meat.
- SOR/78-874, s. 3
- SOR/2018-69, s. 9(F)
B.22.017 [S]. Vegetable Stew with (naming the poultry meat)
(a) shall contain vegetables and the named poultry meat in the following amounts:
(i) if uncooked, 12 per cent or more of the named stew poultry meat,
(ii) if cooked, 6 per cent or more of the named stew poultry meat,
(iii) 38 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 3
B.22.018 [S]. (naming the poultry meat) Stew
(a) shall contain vegetables and the named poultry meat in the following amounts:
(i) if uncooked, 20 per cent or more of the named stew poultry meat,
(ii) if cooked, 10 per cent or more of the named stew poultry meat,
(iii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 3
B.22.019 [S]. Specialty Poultry Meat Stew
(a) shall contain poultry meat and vegetables in the following amounts:
(i) if uncooked, 25 per cent or more of stew poultry meat,
(ii) if cooked, 15 per cent or more of stew poultry meat,
(iii) 30 per cent or more vegetables; and
(b) may contain gravy, salt, seasoning and spices.
- SOR/78-874, s. 3
Prepared Poultry Meats, Prepared Poultry Meat By-Products
B.22.020 [Repealed, SOR/86-875, s. 7]
B.22.021 [S]. Preserved poultry meat or poultry meat by-product shall be cooked or uncooked poultry meat or poultry meat by-product that is cured or smoked and may contain
(a) a Class I preservative;
(a.1) a Class II preservative;
(b) liquid smoke flavour, liquid smoke flavour concentrate or spices;
(c) sweetening agents;
(d) vinegar;
(e) in the case of cured poultry or poultry meat prepared by means of injection or cover solution, disodium phosphate, monosodium phosphate, sodium hexametaphosphate, sodium tripolyphosphate, tetrasodium pyrophosphate and sodium acid pyrophosphate, in such amount calculated as disodium phosphate, as will result in the finished product containing not more than 0.5 per cent added phosphate; and
(f) in the case of vacuum-packed sliced cooked turkey, Carnobacterium maltaromaticum CB1.
- SOR/80-13, s. 13
- SOR/82-596, s. 10
- SOR/94-567, s. 5
- SOR/2010-264, s. 6
- SOR/2011-280, s. 12
- SOR/2017-18, s. 12(F)
- SOR/2018-69, s. 10(F)
B.22.022 [S]. Canned (naming the poultry) shall be prepared from poultry meat and may contain
(a) those bones or pieces of bones attached to the portion of the poultry meat that is being canned;
(b) broth;
(c) salt;
(d) seasoning;
(e) gelling agents; and
(f) small amounts of fat.
- SOR/84-300, s. 55
B.22.023 [S]. Broth that is used in canned (naming the poultry) shall be the liquid in which the poultry meat has been cooked.
B.22.024 [Repealed, SOR/2022-143, s. 27]
B.22.025 [S]. Boneless (naming the poultry) shall be canned poultry meat from which the bones and skin have been removed, shall contain not less than 50 per cent of the named poultry meat, as determined by official method FO-39, Determination of Meat in Boneless Poultry, October 15, 1981, and may contain broth having a specific gravity of not less than 1.000 at a temperature of 50°C.
- SOR/82-768, s. 69
B.22.026 No person shall sell poultry, poultry meat or poultry meat by-product that has been barbecued, roasted or broiled and is ready for consumption unless the cooked poultry, poultry meat or poultry meat by-product
(a) at all times
(i) has a temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher, or
(ii) has been stored at an ambient temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher; and
(b) carries on the principal display panel of the label a statement to the effect that the food must be stored at a temperature of 40°F (4.4°C) or lower, or 140°F (60°C) or higher.
- SOR/78-403, s. 28(F)
- SOR/88-336, s. 3
Poultry Product Extender
B.22.027 No person shall sell a poultry product extender unless that extender
(a) has, in the rehydrated state,
(i) a total protein content of not less than 16 per cent; and
(ii) a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(b) has, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to Division 14 in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(c) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 70
Extended Poultry Products
B.22.028 No person shall sell a food that consists of a mixture of poultry product and poultry product extender, unless that food
(a) has a total protein content of not less than 16 per cent, and
(b) has a protein rating of not less than 40, as determined by official method, and unless
the poultry product extender meets the requirements of paragraphs B.22.027(a) to (c).
Simulated Poultry Products
B.22.029 No person shall sell a simulated poultry product unless that product
(a) has a total protein content of not less than 16 per cent;
(b) has a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) has a fat content of not more than 15 per cent;
(d) contains, notwithstanding sections D.01.009 and D.02.009, each vitamin and mineral nutrient listed in Column I of the Table to Division 14 in an amount not less than the amount shown in Column II of that Table opposite each such vitamin and mineral nutrient respectively; and
(e) where isolated essential amino acids have been added, contains those acids in an amount not exceeding an amount that improves the nutritional quality of the protein.
- SOR/82-768, s. 71
Egg Products
B.22.032 No person shall sell any product simulating whole egg unless that product
(a) is made from liquid, dried or frozen egg albumen or mixtures thereof;
(b) has a protein rating of not less than 40, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981;
(c) notwithstanding sections D.01.009 and D.02.009, contains, per 100 grams on a ready-to-use basis,
(i) not less than
(A) 50 milligrams calcium,
(B) 2.3 milligrams iron,
(C) 1.5 milligrams zinc,
(D) 130 milligrams potassium,
(E) 1000 International Units Vitamin A,
(F) 0.10 milligram thiamine,
(G) 0.30 milligram riboflavin,
(H) 3.60 milligrams niacin,
(I) 1.60 milligrams pantothenic acid,
(J) 0.20 milligram Vitamin B6′,
(K) 0.50 microgram Vitamin B12′,
(L) 0.02 milligram folic acid, and
(M) 2.0 International Units alpha tocopherol, and
(ii) not more than 3 milligrams cholesterol;
(d) has a calcium to phosphorous ratio of not less than one part calcium to four parts phosphorous; and
(e) contains in the total fat of any fat or oil used not less than 40 per cent cis-cis methylene interrupted polyunsaturated fatty acids and not more than 20 per cent saturated fatty acids.
- SOR/82-768, s. 72
- SOR/84-300, s. 56
B.22.033 No person shall sell any egg product referred to in sections B.22.032, B.22.034, B.22.035, B.22.036 and B.22.037 for use as food unless it is free from bacteria of the genus Salmonella, as determined by official method MFO-6, Microbiological Examination of Egg Products and Liquid Eggs, November 30, 1981.
- SOR/82-768, s. 73
B.22.034 [S]. Liquid Whole Egg, Dried Whole Egg or Frozen Whole Egg
(a) shall be the product obtained by removing the shell from wholesome fresh eggs or wholesome stored eggs, and
(i) in the case of dried whole egg, drying the product, or
(ii) in the case of frozen whole egg, freezing the product; and
(b) may
(i) contain aluminum sulphate, pH adjusting agents and the colour beta carotene,
(ii) in the case of liquid whole egg destined for drying, contain yeast autolysate and may be treated with hydrogen peroxide and catalase, glucose oxidase and catalase or yeast and suitable glucose fermenting bacterial culture, or
(iii) in the case of dried whole egg, contain anti-caking agents.
B.22.035 [S]. Liquid Yolk, Dried Yolk or Frozen Yolk
(a) shall be the product obtained by removing the shell and egg-white from wholesome fresh eggs or wholesome stored eggs, and
(i) in the case of dried yolk, drying the product, or
(ii) in the case of frozen yolk, freezing the product, and
(b) may
(i) contain aluminum sulphate, pH adjusting agents and the colour beta carotene,
(ii) in the case of liquid yolk destined for drying, contain yeast autolysate and may be treated with hydrogen peroxide and catalase, glucose oxidase and catalase or yeast and suitable glucose fermenting bacterial culture, or
(iii) in the case of dried yolk, contain anti-caking agents.
B.22.036 [S]. Liquid Egg-White, (Liquid Albumen), Dried Egg-White, (Dried Albumen) or Frozen Egg-White (Frozen Albumen)
(a) shall be the product obtained by removing the shell and yolk from wholesome fresh eggs or wholesome stored eggs, and
(i) in the case of dried egg-white, drying the product, or
(ii) in the case of frozen egg-white, freezing the product; and
(b) may
(i) contain whipping agents, aluminum sulphate and pH adjusting agents,
(ii) in the case of liquid egg-white destined for drying, contain yeast autolysate and may be treated with hydrogen peroxide and catalase, glucose oxidase and catalase or yeast and suitable glucose fermenting bacterial culture,
(iii) in the case of liquid egg-white and dried egg-white, contain lipase or pancreatin, or
(iv) in the case of dried egg-white, contain anti-caking agents.
B.22.037 [S]. Liquid Whole Egg Mix, Dried Whole Egg Mix, Frozen Whole Egg Mix, Liquid Yolk Mix, Dried Yolk Mix or Frozen Yolk Mix
(a) shall be the product obtained by adding salt, sweetening agent or both to Liquid Whole Egg, Dried Whole Egg, Frozen Whole Egg, Liquid Yolk, Dried Yolk or Frozen Yolk; and
(b) may, in the case of dried whole egg mix or dried yolk mix, contain anti-caking agents.
B.22.038 (1) No person shall use a common name referred to in sections B.22.034 to B.22.037 for an egg product that has been subjected to a process, other than a process referred to in those sections, if that process results in a decrease in the amount of a vitamin or mineral nutrient that before processing was present in 100 g of the egg product in an amount equal to at least 10 per cent of the weighted recommended nutrient intake, unless the amount of the vitamin or mineral nutrient is restored to the amount that was present before processing.
(2) Notwithstanding sections D.01.009, D.01.011 and D.02.009, a person may add any vitamin or mineral nutrient referred to in column II of item 27 of the table to section D.03.002 to any egg product referred to in sections B.22.034 to B.22.037 to restore the vitamin or mineral nutrient to the amount that was present in the egg product before processing.
(3) In this section, weighted recommended nutrient intake has the same meaning as in subsection D.01.001(1).
- SOR/96-259, s. 2
DIVISION 23Food Packaging Materials
B.23.001 No person shall sell any food in a package that may yield to its contents any substance that may be injurious to the health of a consumer of the food.
B.23.002 Subject to section B.23.003 no person shall sell any food in a package that has been manufactured from a polyvinyl chloride formulation containing an octyltin chemical.
B.23.003 A person may sell food, other than milk, skim milk, partly skimmed milk, sterilized milk, malt beverages and carbonated non-alcoholic beverage products, in a package that has been manufactured from a polyvinyl chloride formulation containing any or all of the octyltin chemicals, namely, di(n-octyl)tin S,S′-bis(isooctylmercaptoacetate), di(n-octyl)tin maleate polymer and (n-octyl)tin S,S′,S″-tris(isooctylmercaptoacetate) if the proportion of such chemicals, either singly or in combination, does not exceed a total of three per cent of the resin, and the food in contact with the package contains not more than one part per million total octyltin.
- SOR/81-60, s. 13
- SOR/86-1125, s. 4
B.23.004 (1) Di (n-octyl)tin S,S′-bis (isooctylmercaptoacetate) shall be the octyltin chemical made from di (n-octyl)tin dichloride and shall contain 15.1 to 16.4 per cent of tin and 8.1 to 8.9 per cent of mercapto sulfur.
(2) For the purposes of this Division, di (n-octyl)tin dichloride shall be the chemical having an organotin composition of not less than 95 per cent di (n-octyl)tin dichloride and shall contain no more than
(a) five per cent total of n-octyltin trichloride or tri(n-octyl)tin chloride or both;
(b) 0.2 per cent total of other eight (8) carbon isomeric alkyltin derivatives; and
(c) 0.1 per cent total of the higher and lower homologous alkyltin derivatives.
- SOR/86-1125, s. 5
B.23.005 Di(n-octyl)tin maleate polymer shall be the octyltin chemical made from di(n-octyl)tin dichloride and shall have the formula ((C8H17)2 SnC4H2O4)n (where n is between 2 and 4 inclusive), and a saponification number of 225 to 255, and shall contain 25.2 to 26.6 per cent of tin.
- SOR/86-1125, s. 6(F)
B.23.006 (1) (n-octyl)tin S,S′,S″-tris (isooctylmercaptoacetate), being an octyltin chemical having the formula n-C8H17Sn(SCH2CO2C8H17)3, shall be made from (n-octyl)tin trichloride and shall contain 13.4 to 14.8 per cent of tin and 10.9 to 11.9 per cent of mercapto sulfur.
(2) For the purposes of this Division, (n-octyl)tin trichloride shall be the chemical having an organotin composition of not less than 95 per cent (n-octyl)tin trichloride and shall contain not more than
(a) five per cent total of di(n-octyl)tin dichloride, tri(n-octyl)tin chloride or the higher (more than eight (8) carbons) alkyltin chlorides or any combination of the foregoing;
(b) 0.2 per cent total of alkyltin derivatives; and
(c) 0.1 per cent of the lower (less than eight carbons) homologous alkyltin derivatives.
- SOR/86-1125, s. 7
B.23.007 No person shall sell a food in a package than may yield to its contents any amount of vinyl chloride, as determined by official method, FO-40, Determination of Vinyl Chloride in Food, October 15, 1981, in respect of that food.
- SOR/82-768, s. 74
B.23.008 No person shall sell a food in a package that may yield to its contents any amount of acrylonitrile as determined by official method FO-41, Determination of Acrylonitrile in Food, February 16, 1982, in respect of that food.
- SOR/82-541, s. 1
DIVISION 24
Foods for Special Dietary Use
B.24.001 In this Division,
- expiration date
expiration date means, in respect of a formulated liquid diet, a food represented for use in a very low-energy diet, a meal replacement or a nutritional supplement, the date
(a) after which the manufacturer does not recommend that it be consumed, and
(b) up to which it maintains its microbiological and physical stability and the nutrient content declared on the label; (date limite d’utilisation)
- food for special dietary use
food for special dietary use means food that has been specially processed or formulated to meet the particular requirements of a person
(a) in whom a physical or physiological condition exists as a result of a disease, disorder or injury, or
(b) for whom a particular effect, including but not limited to weight loss, is to be obtained by a controlled intake of foods; (aliment à usage diététique spécial)
- formulated liquid diet
formulated liquid diet means a food that
(a) is sold for consumption in liquid form, and
(b) is sold or represented as a nutritionally complete diet for oral or tube feeding of a person described in paragraph (a) of the definition “food for special dietary use”; (préparation pour régime liquide)
- hospital
hospital means a facility
(a) that is licensed, approved or designated as a hospital by a province, in accordance with the laws of the province, to provide care or treatment to persons suffering from any form of disease or illness, or
(b) that is owned or operated by the government of Canada or of a province and that provides health services; (hôpital)
- major change
major change means, in respect of a food that is represented for use in a very low energy diet, any change in any of the following, where the manufacturer’s experience or generally accepted theory would predict an adverse effect on the levels or availability of nutrients in, the microbiological or chemical safety of or the safe use of the food:
(a) an ingredient or the amount of an ingredient in the food,
(b) the manufacturing process or the packaging of the food, or
(c) the directions for the preparation and use of the food; (changement majeur)
- meal replacement
meal replacement[Repealed, SOR/95-474, s. 3]
- pharmacist
pharmacist means a person who is registered and entitled under the laws of a province to practise pharmacy and who is practising pharmacy under those laws in that province; (pharmacien)
- physician
physician means a person who is registered and entitled under the laws of a province to practise medicine and who is practicing medicine under those laws in that province; (médecin)
- prepackaged meal
prepackaged meal[Repealed, SOR/95-474, s. 3]
- target body weight
target body weight means the anticipated body weight at the end of the weight reduction diet, as determined by the physician before the weight reduction diet begins; (poids corporel cible)
- very low energy diet
very low energy diet means a diet for weight reduction that provides less than 900 kilocalories per day when followed as directed. (régime à très faible teneur en énergie)
- SOR/78-64, s. 1
- SOR/78-698, s. 4
- SOR/94-35, s. 1
- SOR/95-474, s. 3
B.24.003 (1) No person shall label, package, sell or advertise a food in a manner likely to create an impression that it is a food for special dietary use unless the food is
(a) to (e) [Repealed, SOR/2003-11, s. 21]
(f) a formulated liquid diet that meets the requirements contained in sections B.24.101 and B.24.102;
(f.1) a meal replacement for special dietary use that meets the requirements contained in section B.24.200;
(f.2) a nutritional supplement that meets the requirements contained in section B.24.201;
(g) a gluten-free food that meets the requirements contained in section B.24.018;
(h) represented for protein-restricted diets;
(i) represented for low (naming the amino acid) diets; or
(j) a food represented for use in a very low energy diet, where the food meets the requirements contained in section B.24.303.
(1.1) Despite subsection (1), a person may label, package, sell or advertise a food in a manner likely to create an impression that it is a food for special dietary use if its label carries a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, in accordance with section B.01.503, in respect of any of the following subjects set out in column 1:
(a) “free of energy”, set out in item 1;
(b) “low in energy”, set out in item 2;
(c) “free of sodium or salt”, set out in item 31;
(d) “low in sodium or salt”, set out in item 32; or
(e) “free of sugars”, set out in item 37.
(2) Subsection (1) does not apply to a human milk fortifier or human milk substitute as defined in section B.25.001.
(3) No person shall label, package, sell or advertise a food in a manner likely to create an impression that it is for use in a weight reduction diet unless that food is
(a) a meal replacement that meets the compositional requirements contained in section B.24.200;
(b) a prepackaged meal;
(c) a food sold by a weight reduction clinic to clients of the clinic for use in a weight reduction program supervised by the staff of the clinic; or
(d) a food represented for use in a very low-energy diet that meets the compositional requirements contained in section B.24.303.
(4) Except as otherwise permitted by these Regulations, no person shall label, package, sell or advertise a food as “dietetic” or “diet”, or use those words as part of the brand name of the food, unless its label carries a statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims, in accordance with section B.01.503, in respect of any of the following subjects set out in column 1:
(a) “free of energy”, set out in item 1;
(b) “low in energy”, set out in item 2;
(c) “reduced in energy”, set out in item 3;
(d) “lower in energy”, set out in item 4; or
(e) “free of sugars”, set out in item 37.
- SOR/78-64, s. 2
- SOR/78-698, s. 5
- SOR/84-334, s. 1
- SOR/86-178, s. 8(E)
- SOR/94-35, s. 2
- SOR/95-444, s. 1
- SOR/95-474, s. 4
- SOR/2003-11, s. 21
- SOR/2021-57, s. 11
- SOR/2022-168, s. 52
B.24.004 to B.24.014 [Repealed, SOR/2003-11, s. 22]
B.24.015 and B.24.016 [Repealed, SOR/88-559, s. 6]
B.24.017 (1) Where the manufacturer of a formulated liquid diet, a meal replacement or a food represented for use in a very low energy diet is requested in writing by the Minister to submit, on or before a specified day, evidence with respect to that product, the manufacturer shall make no further sales of that product after that day unless the manufacturer has submitted the evidence requested.
(2) If the Minister determines that the evidence submitted by a manufacturer under subsection (1) is not sufficient, he or she shall so notify the manufacturer in writing.
(3) Where, pursuant to subsection (2), a manufacturer is notified that the evidence with respect to a formulated liquid diet, a meal replacement or a food represented for use in a very low energy diet is not sufficient, the manufacturer shall make no further sales of that product unless the manufacturer submits further evidence and is notified in writing by the Minister that the further evidence is sufficient.
(4) A reference in this section to evidence with respect to a formulated liquid diet, a meal replacement or a food represented for use in a very low energy diet means evidence to establish that the food is nutritionally adequate to be used as the sole source of nutrition in meeting the nutritional needs of a person for whom it is intended, when the food is consumed in accordance with the directions for use.
- SOR/78-698, s. 6
- SOR/94-35, s. 3
- SOR/2018-69, ss. 11, 27
B.24.018 It is prohibited to label, package, sell or advertise a food in a manner likely to create an impression that it is a gluten-free food if the food contains any gluten protein or modified gluten protein, including any gluten protein fraction, referred to in the definition gluten in subsection B.01.010.1(1).
- SOR/95-444, s. 2
- SOR/2011-28, s. 6
B.24.019 [Repealed, SOR/2003-11, s. 23]
Formulated Liquid Diets
B.24.100 No person shall advertise a formulated liquid diet to the general public.
- SOR/78-64, s. 7
- SOR/78-698, s. 7
B.24.101 No person shall sell a formulated liquid diet unless the food
(a) if sold ready to serve, or
(b) if not sold ready to serve, when diluted with water, milk, or water and milk,
is a complete substitute for the total diet in meeting the nutritional requirements of a person.
- SOR/78-64, s. 7
B.24.102 (1) Subject to subsection (4), a formulated liquid diet shall contain, when ready to serve,
(a) either
(i) not less than 20 grams of protein of nutritional quality equivalent to casein, as determined by official method FO-1, Determination of Protein Rating, October 15, 1981, or
(ii) such an amount and quality of protein, including those proteins to which amino acids are added, that, when the quality of the protein is expressed as a fraction of the quality of casein,
(A) the fraction will not be less than 85/100, and
(B) the result obtained by multiplying the fraction by the gram weight of the protein will not be less than 20; and
(b) not less than one gram linoleic acid in the form of a glyceride.
(2) Notwithstanding sections D.01.009, D.01.011 and D.02.009, a formulated liquid diet shall contain, when ready to serve, the vitamins and minerals named in Column I of the table to this section in amounts,
(a) where the recommended intake of the food is 2,500 kilocalories per day or less, not less than the amounts set out in Column II and not more than the amounts, if any, set out in Column III of that table opposite those vitamins and minerals; and
(b) where the recommended intake of the food is greater than 2,500 kilocalories per day, not less than the amounts set out in Column IV and not more than the amounts, if any, set out in Column V of the table opposite those vitamins and minerals.
(3) The amounts of the nutrients specified in paragraphs (1)(a) and (b) and subsection (2) shall be calculated
(a) per 1,000 available kilocalories, where the recommended intake of the food is 2,500 kilocalories per day or less; and
(b) per 1,500 available kilocalories, where recommended intake of the food is greater than 2,500 kilocalories per day.
(4) Paragraph (1)(a) does not apply to a formulated liquid diet represented as being for a protein restricted diet or a low (named amino acid) diet.
TABLE
Column I Per 1,000 available kilocalories Per 1,500 available kilocalories Column II Column III Column IV Column V Vitamins Minimum Maximum Minimum Maximum Vitamin A 2,000 I.U. 5,000 I.U. 2,000 I.U. 3,000 I.U. Vitamin D 100 I.U. 400 I.U. 100 I.U. 200 I.U. Vitamin E (a-tocopherol) 5.0 I.U. 5.0 I.U. Ascorbic Acid 20 mg 20 mg Thiamine 0.5 mg 0.6 mg Riboflavin 0.7 mg 0.84 mg Niacin 6.6 mg 7.9 mg Vitamin B6 0.9 mg 0.9 mg Vitamin B12 1.5 µg 1.5 µg Folic Acid 100 µg 100 µg d-pantothenic Acid 2.5 mg 2.5 mg Mineral Nutrients Calcium 400 mg 400 mg Phosphorus 400 mg 400 mg Iron 8 mg 8 mg Iodine 50 µg 50 µg Magnesium 150 mg 150 mg Copper 1 mg 1 mg Zinc 7 mg 7 mg
- SOR/78-64, s. 7
- SOR/78-698, s. 8
- SOR/82-768, s. 75
- SOR/87-640, s. 9(F)
- SOR/90-830, s. 6(F)
- SOR/2022-197, s. 5(E)
B.24.103 The label of a formulated liquid diet shall carry the following information:
(a) a statement that the food is intended to be consumed orally or by tube feeding;
(b) a statement of the energy value of the food, expressed in Calories
(i) per 100 grams or per 100 millilitres of the food as offered for sale, and
(ii) per unit of ready-to-serve food;
(c) a statement of the content in the food of protein or protein equivalent, fat, linoleic acid, available carbohydrate and, where present, crude fibre, expressed in grams
(i) per 100 grams or per 100 millilitres of the food as offered for sale, and
(ii) per unit of ready-to-serve food;
(d) a statement of the content of vitamins and mineral nutrients that are listed in the table to section B.24.102, expressed in International Units, milligrams or micrograms
(i) per 100 grams or per 100 millilitres of the food as offered for sale, and
(ii) per unit of ready-to-serve food;
(e) a statement of the content of any vitamin or mineral nutrient that is not listed in the table to section B.24.102, expressed in milligrams or micrograms
(i) per 100 grams or per 100 millilitres of the food as offered for sale, and
(ii) per unit of ready-to-serve food;
(f) complete directions for the preparation and use of the food and for its storage after the container has been opened; and
(g) the expiration date of the formulated liquid diet.
- SOR/78-64, s. 7
- SOR/88-559, s. 27
- SOR/2022-197, s. 6
Meal Replacements, Nutritional Supplements, Prepackaged Meals and Foods Sold by Weight Reduction Clinics
B.24.200 (1) No person shall sell or advertise a meal replacement unless, when in a ready-to-serve form or when prepared according to directions for use, with water, milk, partially skim milk or skim milk, or a combination thereof, it meets the following requirements:
(a) the meal replacement provides a minimum of 225 kcal or 945 kJ per serving;
(b) not less than 15 per cent and not more than 40 per cent of the energy available from the meal replacement is derived from its protein content, except that a meal replacement for use in a weight reduction diet shall derive not less than 20 per cent of its available energy from its protein content;
(c) subject to subsection (2), not more than 35 per cent of the energy available from the meal replacement is derived from its fat content;
(d) not less than 3.0 per cent of the energy available from the meal replacement is derived from linoleic acid in the form of a glyceride and not less than 0.5 per cent of the energy available from the meal replacement is derived from n-3 linolenic acid in the form of a glyceride, and the ratio of linoleic acid to n-3 linolenic acid is not less than 4 to 1 and not more than 10 to 1;
(e) the proteins present in the meal replacement are
(i) of a nutritional quality equivalent to that of casein, or
(ii) of a nutritional quality and in an amount sufficient to yield a result of not less than 15 per cent, or not less than 20 per cent in the case of a meal replacement for use in a weight reduction diet, when the nutritional quality of those proteins is divided by the nutritional quality of casein and multiplied by the percentage of energy available from the proteins present in the meal replacement; and
(f) each serving of the meal replacement contains each vitamin and mineral nutrient listed in column I of the table to this section
(i) subject to subsection (3), in an amount not less than the minimum amount shown for that vitamin or mineral nutrient in column II of the table, and
(ii) subject to subsections (4) and (5), in an amount that, including overage, is not more than the maximum amount shown for that vitamin or mineral nutrient in column III of the table.
(2) No person shall sell or advertise a meal replacement that is represented as a replacement for all daily meals unless, when in a ready-to-serve form or when prepared according to directions for use, with water, milk, partially skim milk or skim milk, or a combination thereof, it meets the following requirements:
(a) not more than 30 per cent of the energy available from the meal replacement is derived from its fat content; and
(b) not more than 10 per cent of the energy available from the meal replacement is derived from its saturated fatty acid content.
(3) The minimum amount required under subparagraph (1)(f)(i) for selenium, chromium or molybdenum does not apply in respect of a meal replacement that is not represented as a replacement for all daily meals and that does not contain added selenium, chromium or molybdenum, as the case may be.
(4) A vitamin or mineral nutrient that is not an added ingredient in the meal replacement shall not be taken into account for the purposes of subparagraph (1)(f)(ii).
(5) The maximum amount shown for vitamin C in column III of the table to this section does not include overage.
TABLE
Column I Column II Column III Nutrients Minimum Amount per Serving Maximum Amount per Serving VITAMINS Vitamin A 250 RE 630 RE Vitamin D 1.25 µg 2.50 µg Vitamin E 2.5 mg 5.0 mg Vitamin C 10 mg 20 mg Thiamine 300 µg 750 µg Riboflavin 400 µg 800 µg Niacin 6 NE 12 NE Vitamin B6 400 µg 750 µg Vitamin B12 0.25 µg 0.75 µg Folacin 60 µg 120 µg Pantothenic acid 1.25 mg 2.50 mg Biotin 25 µg 75 µg MINERAL NUTRIENTS Calcium 200 mg 400 mg Phosphorus 250 mg 500 mg Iron 2.5 mg 5.0 mg Iodide 40 µg 120 µg Magnesium 60 mg 120 mg Copper 0.5 mg 1.0 mg Zinc 3 mg 6 mg Potassium 375 mg Sodium 250 mg Manganese 1 mg 2 mg Selenium 10 µg 20 µg Chromium 10 µg 20 µg Molybdenum 20 µg 40 µg
- SOR/78-698, s. 9
- SOR/80-13, s. 14
- SOR/95-474, s. 5
B.24.201 (1) No person shall sell or advertise a nutritional supplement that contains less than 225 kcal or 945 kJ per serving, unless it meets the following requirements:
(a) the nutritional supplement contains at least 150 kcal or 630 kJ per serving;
(b) not less than 15 per cent and no more than 40 per cent of the energy available from the nutritional supplement is derived from its protein content;
(c) the proteins present in the nutritional supplement are
(i) of a nutritional quality equivalent to that of casein, or
(ii) of a nutritional quality and in an amount sufficient to yield a result of not less than 15 per cent when the nutritional quality of those proteins is divided by the nutritional quality of casein and multiplied by the percentage of energy available from the proteins present in the nutritional supplement; and
(d) the nutritional supplement contains, per 100 kcal or 420 kJ, each vitamin and mineral nutrient listed in column I of the table to this section
(i) subject to subsection (3), in an amount not less than the minimum amount shown for that vitamin or mineral nutrient in column II of the table, and
(ii) subject to subsections (4) and (5), in an amount that, including overage, is not more than the maximum amount shown for that vitamin or mineral nutrient in column III of the table.
(2) No person shall sell or advertise a nutritional supplement that provides 225 kcal or 945 kJ, or more, per serving unless, when in a ready-to-serve form or when prepared according to directions for use, with water, milk, partially skim milk, skim milk, or a combination thereof, it meets the following requirements:
(a) the nutritional supplement provides at least 225 kcal or 945 kJ per serving;
(b) not more than 35 per cent of the energy available from the nutritional supplement is derived from its fat content;
(c) not less than 3.0 per cent of the energy available from the nutritional supplement is derived from linoleic acid in the form of a glyceride and not less than 0.5 per cent of the energy available from the nutritional supplement is derived from n-3 linolenic acid in the form of a glyceride, and the ratio of linoleic acid to n-3 linolenic acid is not less than 4 to 1 and not more than 10 to 1;
(d) not less than 15 per cent and not more than 40 per cent of the energy available from the nutritional supplement is derived from its protein content;
(e) the proteins present in the nutritional supplement are
(i) of a nutritional quality equivalent to that of casein, or
(ii) of a nutritional quality and in an amount sufficient to yield a result of not less than 15 per cent when the nutritional quality of those proteins is divided by the nutritional quality of casein and multiplied by the percentage of energy available from the proteins present in the nutritional supplement; and
(f) the nutritional supplement contains, per 100 kcal or 420 kJ, each vitamin and mineral nutrient listed in column I of the table to this section
(i) subject to subsection (3), in an amount not less than the minimum amount shown for that vitamin or mineral nutrient in column II of the table, and
(ii) subject to subsections (4) and (5), in an amount that, including overage, is not more than the maximum amount shown for that vitamin or mineral nutrient in column III of the table.
(3) The minimum amount required under subparagraph (1)(d)(i) or (2)(f)(i) for selenium, chromium or molybdenum does not apply in respect of a nutritional supplement that does not contain added selenium, chromium or molybdenum, as the case may be.
(4) A vitamin or mineral nutrient that is not an added ingredient in the nutritional supplement shall not be taken into account for the purposes of subparagraphs (1)(d)(ii) and (2)(f)(ii).
(5) The maximum amount shown for vitamin C in column III of the table to this section does not include overage.
TABLE
Column I Column II Column III Nutrients Minimum Amount per Available 100 Kcal or 420 KJ Maximum Amount per Available 100 Kcal or 420 KJ VITAMINS Vitamin A 100 RE 250 RE Vitamin D 0.25 µg 1 µg Vitamin E 1.0 mg 2.0 mg Vitamin C 5 mg 10 mg Thiamine 140 µg 350 µg Riboflavin 180 µg 360 µg Niacin 3 NE 6 NE Vitamin B6 180 µg 350 µg Vitamin B12 0.1 µg 0.3 µg Folacin 30 µg 60 µg Pantothenic acid 0.6 mg 1.2 mg Biotin 12 µg 35 µg MINERAL NUTRIENTS Calcium 100 mg 175 mg Phosphorus 100 mg 175 mg Iron 1.0 mg 2.0 mg Iodide 15 µg 45 µg Magnesium 20 mg 40 mg Copper 0.15 mg 0.30 mg Zinc 1.4 mg 2.0 mg Potassium 175 mg Manganese 0.45 mg 0.90 mg Selenium 4 µg 8 µg Chromium 4 µg 8 µg Molybdenum 8 µg 15 µg
- SOR/78-698, s. 9
- SOR/95-474, s. 5
B.24.202 The label of a meal replacement or nutritional supplement shall
(a) show the following information per serving of stated size and per stated quantity of food, when prepared according to the directions for use:
(i) the energy value of the food, expressed in Calories (Calories or Cal) and kilojoules (kilojoules or kJ),
(ii) the protein, fat, linoleic acid, n-3 linolenic acid, saturated fatty acid and carbohydrate contents of the food, expressed in grams,
(iii) the vitamin A, vitamin D, vitamin E, vitamin C, thiamin or vitamin B1, riboflavin or vitamin B2, niacin, vitamin B6, vitamin B12, folate and pantothenic acid or pantothenate contents of the food, expressed
(A) in the case of a meal replacement, as a percentage of the daily value specified in column 4 of Part 2 of the Table of Daily Values for that vitamin, and
(B) in the case of a nutritional supplement, in the applicable unit referred to in subsection D.01.003(1),
(iv) the calcium, phosphorus, iron, iodide, magnesium and zinc contents of the food, expressed
(A) in the case of a meal replacement, as a percentage of the daily value specified in column 4 of Part 2 of the Table of Daily Values, and
(B) in the case of a nutritional supplement, in milligrams for calcium, phosphorus, iron, magnesium and zinc and in micrograms for iodide,
(v) the copper, potassium, sodium and manganese contents of the food, expressed in milligrams, and
(vi) the biotin, selenium, chromium and molybdenum contents of the food, expressed in micrograms;
(b) in the case of a meal replacement or a nutritional supplement to which milk, partially skim milk or skim milk is to be added, carry a statement that the nutrient content of the food has been determined taking into consideration the milk, partially skim milk or skim milk that will be added according to the directions for use;
(c) in the case of a meal replacement that is sold or advertised as a replacement for all daily meals in a weight reduction diet, include directions for use that would result in a daily energy intake of at least 900 kcal or 3 780 kJ;
(d) include the expiration date of the meal replacement or nutritional supplement;
(e) in the case of a meal replacement for use in a weight reduction diet, carry the statement “USEFUL IN WEIGHT REDUCTION ONLY AS PART OF AN ENERGY-REDUCED DIET / UTILE POUR PERDRE DU POIDS SEULEMENT DANS LE CADRE D’UN RÉGIME À TENEUR RÉDUITE EN ÉNERGIE” prominently displayed on the principal display panel; and
(f) in the case of a meal replacement for use in a weight reduction diet that is not represented as a replacement for all daily meals in a diet, include the information required under section B.24.204.
- SOR/78-698, s. 9
- SOR/88-559, s. 28
- SOR/95-474, s. 5
- SOR/2016-305, ss. 59, 75(F)
B.24.203 The label of a prepackaged meal for use in a weight reduction diet or of a food to be sold in a weight reduction clinic shall
(a) [Repealed, SOR/2003-11, s. 24]
(b) carry the statement “USEFUL IN WEIGHT REDUCTION ONLY AS PART OF AN ENERGY-REDUCED DIET / UTILE POUR PERDRE DU POIDS SEULEMENT DANS LE CADRE D’UN RÉGIME À TENEUR RÉDUITE EN ÉNERGIE” prominently displayed on the principal display panel; and
(c) include the information required under section B.24.204.
- SOR/78-698, s. 9
- SOR/88-559, s. 29
- SOR/95-474, s. 5
- SOR/2003-11, s. 24
B.24.204 The label of a prepackaged meal, or of a meal replacement other than a meal replacement represented as a replacement for all daily meals in a diet, that is packaged, sold or advertised for use in a weight reduction diet, or of a food to be sold in a weight reduction clinic, shall include, in the directions for use, a sample seven-day menu in which the prepackaged meal, meal replacement or food is used and which meets the following requirements:
(a) each daily meal includes a minimum of one serving, as described in Canada’s Food Guide to Healthy Eating, published in 1992 by the Department of Supply and Services by authority of the Minister of National Health and Welfare, of one food from each of the following groups:
(i) milk, milk products or their alternatives,
(ii) meat and meat alternatives,
(iii) bread and grain products, and
(iv) vegetables and fruit;
(b) the daily energy intake provided for is not less than 1 200 kcal or 5 040 kJ;
(c) not more than 30 per cent of the total daily energy intake of the seven-day menu is derived from its fat content and not more than 10 per cent of the total daily energy intake of the menu is derived from its saturated fatty acid content;
(d) the mean daily intake of each nutrient listed in column I of the table to this section is not less than the amount shown in column II, in the case of a menu recommended for men, or in column III, in the case of a menu recommended for women; and
(e) the menu does not include any reference to vitamin or mineral supplements.
TABLE
Column I Column II Column III Nutrients Mean Daily Intake Men Women Protein 65 g 55 g VITAMINS Vitamin A 1000 RE 800 RE Vitamin D 5 µg 5 µg Vitamin E 10 mg 7 mg Vitamin C 40 mg 30 mg Thiamin 1 mg 1 mg Riboflavin 1 mg 1 mg Niacin 14 NE 14 NE Vitamin B6 1.5 mg 1.5 mg Vitamin B12 1 µg 1 µg Folacin 230 µg 200 µg Pantothenic Acid 5 mg 5 mg MINERAL NUTRIENTS Calcium 800 mg 800 mg Phosphorus 1000 mg 850 mg Iron 9 mg 13 mg Iodide 160 µg 160 µg Magnesium 250 mg 210 mg Copper 2 mg 2 mg Zinc 12 mg 9 mg
- SOR/78-698, s. 9
- SOR/95-474, s. 5
B.24.205 (1) No person shall label, package, sell or advertise a prepackaged meal or meal replacement for use in a weight reduction diet, or a food to be sold in a weight reduction clinic, in a manner likely to create an impression that consumption of a vitamin or mineral supplement must be part of a weight reduction diet.
(2) No person shall, on a label of or in an advertisement for a prepackaged meal or meal replacement for use in a weight reduction diet, or a food to be sold in a weight reduction clinic, make any direct or indirect reference to a vitamin or mineral supplement.
(3) Every person who advertises a prepackaged meal or meal replacement for use in a weight reduction diet, or a food to be sold in a weight reduction clinic, shall include in the advertisement the statement “USEFUL IN WEIGHT REDUCTION ONLY AS PART OF AN ENERGY-REDUCED DIET / UTILE POUR PERDRE DU POIDS SEULEMENT DANS LE CADRE D’UN RÉGIME À TENEUR RÉDUITE EN ÉNERGIE”.
- SOR/78-698, s. 9
- SOR/95-474, s. 5
Foods Represented for Use in Very Low Energy Diets
B.24.300 No person shall advertise to the general public a food represented for use in a very low energy diet.
- SOR/94-35, s. 4
B.24.301 (1) No person shall sell, without a written order from a physician, a food represented for use in a very low energy diet.
(2) Notwithstanding subsection (1), a person may sell, without a written order from a physician, a food represented for use in a very low energy diet to
(a) a physician;
(b) a wholesale druggist;
(c) a pharmacist; or
(d) a hospital.
(3) No person other than a pharmacist shall sell to the general public a food represented for use in a very low energy diet.
- SOR/94-35, s. 4
B.24.302 A pharmacist shall retain the written order of a physician for a food represented for use in a very low energy diet for at least two years after the date on which the order is filled.
- SOR/94-35, s. 4
B.24.303 (1) A food represented for use in a very low energy diet, whether ready to serve or diluted with water according to the manufacturer’s directions, shall provide, per daily allowance recommended by the manufacturer
(a) either
(i) not less than 60 g of protein of a nutritional quality equivalent to that of casein, or
(ii) such an amount and quality of protein that, when the quality of the protein is expressed as a fraction of the quality of casein,
(A) the fraction is not less than 85/100, and
(B) the product obtained by multiplying the fraction by the gram weight of the protein is not less than 60;
(b) each vitamin or mineral nutrient named in column I of an item of the table to this subsection, in an amount not less than the minimum amount per day set out in column II of that item; and
(c) any nutritive substance added to the food other than those referred to in paragraph (a) or (b), in an amount that is appropriate for the purpose of the substance in the food as determined from clinical trials.
TABLE
Column I Column II Item Vitamin or Mineral Nutrient Minimum amount per day 1 Thiamine 1.3 mg 2 Riboflavin 1.6 mg 3 Niacin 23 mg 4 Folacin 0.22 mg 5 Biotin 0.15 mg 6 Pantothenic acid 7.0 mg 7 Vitamin B6 1.5 mg 8 Vitamin B12 0.001 mg 9 Vitamin A 1000 RE 10 Vitamin D 0.005 mg 11 Vitamin E 10 mg 12 Vitamin C 40 mg 13 Calcium 800 mg 14 Phosphorus 1000 mg 15 Magnesium 250 mg 16 Iron 13 mg 17 Iodine 0.16 mg 18 Zinc 12 mg 19 Copper 2 mg 20 Manganese 3.5 mg 21 Selenium 0.07 mg 22 Chromium 0.05 mg 23 Molybdenum 0.1 mg 24 Sodium 2000 mg 25 Potassium 3000 mg 26 Chloride 1500 mg
(2) Notwithstanding paragraph (1)(a), a food represented for use in a very low energy diet shall be accompanied by directions for use that when followed would result in the daily intake by a person of at least 1.2 g of protein per kilogram target body weight.
- SOR/94-35, s. 4
B.24.304 The label of a food represented for use in a very low energy diet shall carry the following information:
(a) a statement of the energy value of the food, expressed in Calories (Calories or Cal) and kilojoules (kilojoules or kJ) per 100 g or 100 mL of the food as offered for sale and per unit of ready-to-serve food;
(b) a statement of the content in the food of protein, fat, carbohydrate and, where present, fibre expressed in grams per 100 g or 100 mL of the food as offered for sale and per unit of ready-to-serve food;
(c) a statement of the content in the food of all those vitamins and mineral nutrients that are listed in the table to subsection B.24.303(1) expressed in milligrams, in the case of vitamin A expressed in retinol equivalents (RE), per 100 g or 100 mL of the food as offered for sale and per unit of ready-to-serve food;
(d) a statement of the content in the food of any other nutritive substance added to the food in an amount described in paragraph B.24.303(1)(c), expressed in milligrams or in grams per 100 g or 100 mL of the food as offered for sale and per unit of ready-to-serve food;
(e) the statement “USE ONLY UNDER MEDICAL SUPERVISION” prominently displayed on the principal display panel;
(f) directions for use of the food, including
(i) a statement of the rationale for the use of the food,
(ii) criteria to be used for the selection of the persons to whom the food may be prescribed,
(iii) instructions for consultation with and evaluation of the patient and patient follow-up, and
(iv) a statement concerning adequate precautions and contra-indications;
(g) directions for the preparation of the food, and storage instructions for the food before and after the container has been opened; and
(h) the expiration date of the food.
- SOR/94-35, s. 4
B.24.305 (1) No person shall sell or advertise for sale a food represented for use in a very low energy diet unless the manufacturer, at least 90 days before the sale or advertisement, notifies the Minister in writing of the intention to sell the food or advertise the food for sale.
(2) The notification referred to in subsection (1) shall be signed by the manufacturer and shall include, in respect of the food represented for use in a low energy diet, the following information:
(a) the name under which the food is to be sold or advertised for sale;
(b) the name and address of the principal place of business of the manufacturer;
(c) the name and address of each establishment in which the food is manufactured;
(d) a list of the ingredients of the food, stated quantitatively;
(e) the specifications for nutrient, microbiological and physical quality for each ingredient and for the food;
(f) details of quality control procedures respecting the testing of the ingredients and of the food;
(g) details of the manufacturing process and quality control procedures used throughout the process;
(h) the results of tests carried out to determine the expiration date of the food;
(i) the evidence relied on to establish that the food meets the nutritional requirements, other than energy requirements, of a person for whom it is intended, when the food is consumed in accordance with the directions for use;
(j) a description of the type of packaging to be used;
(k) directions for use;
(l) the written text of all labels, including package inserts, to be used in connection with the food; and
(m) the name and title of the person who signed the notification and the date of signature.
(3) Notwithstanding subsection (1), a person may sell or advertise for sale a food represented for use in a very low energy diet, if the Minister, after having been notified by the manufacturer pursuant to that subsection, has informed the manufacturer in writing that the notification meets the requirements of subsection (2).
- SOR/94-35, s. 4
- SOR/2018-69, ss. 27, 29(F)
B.24.306 (1) No person shall sell or advertise for sale a food represented for use in a very low energy diet that has undergone a major change, unless the manufacturer, at least 90 days before the sale or advertisement, notifies the Minister in writing of the intention to sell or advertise for sale the food that has undergone the major change.
(2) The notification referred to in subsection (1) shall be signed by the manufacturer and shall include, in respect of the food represented for use in a very low energy diet that has undergone a major change, the following information:
(a) the name under which the food is to be sold or advertised for sale;
(b) the name and address of the principal place of business of the manufacturer;
(c) a description of the major change;
(d) the evidence relied on to establish that the food meets the nutritional requirements, other than energy requirements, of a person for whom it is intended, when the food is consumed in accordance with the directions for use;
(e) the evidence relied on to establish that the major change has no adverse effect on the food or its use;
(f) the written text of all labels, including package inserts, to be used in connection with the food; and
(g) the name and title of the person who signed the notification and the date of signature.
(3) Notwithstanding subsection (1), a person may sell or advertise for sale a food represented for use in a very low energy diet that has undergone a major change, if the Minister, after having been notified by the manufacturer pursuant to that subsection, has informed the manufacturer in writing that the notification meets the requirements of subsection (2).
- SOR/94-35, s. 4
- SOR/2018-69, ss. 27, 29(F)
DIVISION 25
Interpretation
B.25.001 In this Division,
- expiration date
expiration date means, in respect of a human milk fortifier or human milk substitute, the date
(a) after which the manufacturer does not recommend that it be consumed, and
(b) up to which it maintains its microbiological and physical stability and the nutrient content declared on the label; (date limite d’utilisation)
- human milk fortifier
human milk fortifier means a food that
(a) includes at least one added vitamin, mineral nutrient or amino acid, and
(b) is labelled or advertised as intended to be added to human milk to increase its nutritional value in order to meet the particular requirements of an infant in whom a physical or physiological condition exists as a result of a disease, disorder or abnormal physical state; (fortifiant pour lait humain)
- human milk substitute
human milk substitute means any food that is labelled or advertised
(a) for use as a partial or total replacement for human milk and as intended for consumption by infants, or
(b) for use as an ingredient in a food referred to in paragraph (a); (succédané de lait humain)
- infant
infant means an individual who is under the age of one year; (bébé)
- infant food
infant food means a food that is labelled or advertised for consumption by infants; (aliment pour bébés)
- junior (naming a food)
junior (naming a food) means the named food where it contains particles of a size to encourage chewing by infants, but may be readily swallowed by infants without chewing; ((nom d’un aliment) pour enfants en bas âge)
- major change
major change means, in respect of a human milk fortifier or human milk substitute, any change of an ingredient, the amount of an ingredient or the processing or packaging of the human milk fortifier or human milk substitute where the manufacturer’s experience or generally accepted theory would predict an adverse effect on the levels or availability of nutrients in, or the microbiological or chemical safety of, the human milk fortifier or human milk substitute; (changement majeur)
- new human milk substitute
new human milk substitute means a human milk substitute that is
(a) manufactured for the first time,
(b) sold in Canada for the first time, or
(c) manufactured by a person who manufactures it for the first time; (succédané de lait humain nouveau)
- strained (naming a food)
strained (naming a food) means the named food where it is of a generally uniform particle size that does not require and does not encourage chewing by infants before being swallowed. ((nom d’un aliment) en purée ou tamisé)
- SOR/78-637, s. 5
- SOR/83-933, s. 1
- SOR/90-174, s. 1
- SOR/2021-57, s. 12
Infant Foods
B.25.002 No person shall sell or advertise for sale an infant food that is set out in Column I of an item of Table I to this Division and contains more than the amount of sodium set out in Column II of that item.
- SOR/83-933, s. 1
B.25.003 (1) Subject to subsection (2), no person shall sell infant food that contains
(a) strained fruit,
(b) fruit juice,
(c) fruit drink, or
(d) cereal,
if sodium chloride has been added to that food.
(2) Subsection (1) does not apply to strained desserts containing any of the foods mentioned in paragraphs (1)(a) to (d).
- SOR/83-933, s. 1
Human Milk Fortifiers
B.25.010 Subject to section B.25.013, it is prohibited to sell or advertise for sale a human milk fortifier unless the Minister has notified the manufacturer under paragraph B.25.012(1)(a) or (3)(a) that those activities are authorized.
B.25.011 An application to sell or advertise for sale a human milk fortifier shall be submitted by the manufacturer and signed by the manufacturer or an individual authorized to sign on their behalf and shall include the following information:
(a) the brand name and product name under which the human milk fortifier will be sold or advertised for sale;
(b) the manufacturer’s name and address;
(c) the name and address of each establishment in which the human milk fortifier is manufactured;
(d) a list of all of the human milk fortifier’s ingredients, stated quantitatively;
(e) the scientific rationale for the formulation of the human milk fortifier;
(f) the specifications for nutrient, microbiological and physical quality for the human milk fortifier and its ingredients;
(g) details of the quality control procedures respecting the testing of the human milk fortifier and its ingredients;
(h) details of the human milk fortifier’s manufacturing process and the quality control procedures used throughout the process;
(i) the results of the tests carried out to determine the expiration date of the human milk fortifier;
(j) the evidence that establishes that the human milk fortifier is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use;
(k) a description of the type of packaging to be used for the human milk fortifier;
(l) directions for use for the human milk fortifier;
(m) the written text of all labels, including package inserts, to be used in connection with the human milk fortifier; and
(n) the name and title of the individual who signed the application and the date of signature.
B.25.012 (1) After having conducted an assessment of the information submitted under section B.25.011 and any other information related to the assessment of the application, the Minister shall notify the manufacturer in writing that the sale or advertisement for sale of the human milk fortifier that is the subject of the application
(a) is authorized if
(i) the application meets the requirements set out in section B.25.011, and
(ii) the information is sufficient to establish the safety of the human milk fortifier; or
(b) is not authorized, in any other case.
(2) If the conditions set out in paragraph (1)(a) are not satisfied, the Minister may request the manufacturer to submit in writing the additional information that is necessary to satisfy the conditions.
(3) After having conducted an assessment of any additional information that is submitted by the manufacturer, the Minister shall notify the manufacturer in writing that the sale or advertisement for sale of the human milk fortifier
(a) is authorized, if the conditions set out in paragraph (1)(a) are satisfied; or
(b) is not authorized, in any other case.
(4) Despite paragraphs (1)(a) and (3)(a), the Minister shall not authorize the sale or advertisement for sale of the human milk fortifier if the Minister has reasonable grounds to believe that any of the information that the manufacturer has submitted is false, misleading or deceptive.
B.25.013 The prohibition set out in section B.25.010 does not apply in respect of a human milk fortifier that is set out in the List of Human Milk Fortifiers Sold in Canada as of March 25, 2021 that is published on a Government of Canada website if, on or before April 30, 2022,
(a) the manufacturer of the human milk fortifier submits to the Minister
(i) an attestation signed and dated by the manufacturer, or by an individual authorized to sign on their behalf, confirming that the human milk fortifier has not undergone a major change since the date set out in the List in connection with the human milk fortifier, and
(ii) the written text of all labels, including package inserts, that will be required to be used in connection with the human milk fortifier after the second anniversary of the day on which section B.25.020 comes into force; and
(b) the Minister notifies the manufacturer under subsection B.25.014(1) or paragraph B.25.014(3)(a) that they are authorized to continue to sell or advertise for sale the human milk fortifier.
B.25.014 (1) The Minister shall notify the manufacturer in writing that they are authorized to continue to sell or advertise for sale the human milk fortifier if
(a) the manufacturer has submitted the information referred to in paragraph B.25.013(a) in sufficient time for the Minister to assess the information before April 30, 2022; and
(b) the written text of the labels submitted under subparagraph B.25.013(a)(ii) satisfies the relevant requirements of these Regulations.
(2) If the information that is submitted is insufficient to satisfy the conditions set out in subsection (1), the Minister may request the manufacturer to submit in writing the additional information that is necessary to satisfy the conditions.
(3) After having conducted an assessment of any additional information that is submitted by the manufacturer, the Minister shall notify the manufacturer in writing that
(a) they are authorized to continue to sell or advertise for sale the human milk fortifier, if the conditions set out in subsection (1) are satisfied; or
(b) they are not authorized to continue to sell or advertise for sale the human milk fortifier, in any other case.
B.25.015 (1) It is prohibited to sell or advertise for sale a human milk fortifier that has undergone a major change unless
(a) the manufacturer of the human milk fortifier submits to the Minister an application in accordance with subsection (2) that reflects the major change; and
(b) the Minister notifies the manufacturer under paragraph B.25.016(1)(a) or (3)(a) that the sale or advertisement for sale of the human milk fortifier is authorized.
(2) The application shall be signed by the manufacturer or an individual authorized to sign on their behalf and shall include the following information:
(a) the brand name and product name under which the human milk fortifier will be sold or advertised for sale;
(b) the manufacturer’s name and address;
(c) a description of, and the rationale for, the major change;
(d) the evidence that establishes that the human milk fortifier is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use;
(e) the evidence that establishes that the major change has had no adverse effect on the human milk fortifier;
(f) the written text of all labels, including package inserts, to be used in connection with the human milk fortifier; and
(g) the name and title of the individual who signed the application and the date of signature.
B.25.016 (1) After having conducted an assessment of the information submitted under subsection B.25.015(2) and any other information related to the assessment of the application, the Minister shall notify the manufacturer in writing that the sale or advertisement for sale of the human milk fortifier that is the subject of the application
(a) is authorized if
(i) the application meets the requirements set out in subsection B.25.015(2), and
(ii) the information is sufficient to establish the safety of the human milk fortifier; or
(b) is not authorized, in any other case.
(2) If the conditions set out in paragraph (1)(a) are not satisfied, the Minister may request the manufacturer to submit in writing the additional information that is necessary to satisfy the conditions.
(3) After having conducted an assessment of any additional information that is submitted by the manufacturer, the Minister shall notify the manufacturer in writing that the sale or advertisement for sale of the human milk fortifier
(a) is authorized, if the conditions set out in paragraph (1)(a) are satisfied; or
(b) is not authorized, in any other case.
(4) Despite paragraphs (1)(a) and (3)(a), the Minister shall not authorize the sale or advertisement for sale of the human milk fortifier if the Minister has reasonable grounds to believe that any of the information that the manufacturer has submitted is false, misleading or deceptive.
B.25.017 A manufacturer named in the List of Human Milk Fortifiers Sold in Canada as of March 25, 2021 referred to in section B.25.013 is not entitled to submit an application referred to in paragraph B.25.015(1)(a) in respect of a human milk fortifier set out in the List unless the Minister has
(a) notified them under paragraph B.25.012(1)(a) or (3)(a) that the sale or advertisement for sale of the human milk fortifier is authorized; or
(b) notified them under subsection B.25.014(1) or paragraph B.25.014(3)(a) that they are authorized to continue to sell or advertise for sale the human milk fortifier.
B.25.018 (1) If the manufacturer of a human milk fortifier is requested in writing by the Minister to submit evidence with respect to the human milk fortifier within a time limit specified by the Minister, the manufacturer shall make no further sales of the human milk fortifier — and shall not advertise it for sale — after the expiry of the time limit unless they have submitted the requested evidence.
(2) The time limit cannot be less than 24 hours after the request is made unless the Minister has reasonable grounds to believe that there is a serious and imminent risk of injury to human health.
(3) If the Minister determines that the evidence submitted by the manufacturer is insufficient, the Minister shall notify the manufacturer accordingly in writing.
(4) If the manufacturer is notified that the evidence with respect to the human milk fortifier is insufficient, the manufacturer shall make no further sales of the human milk fortifier – and shall not advertise it for sale — unless they submit additional evidence and are notified in writing by the Minister that the additional evidence is sufficient.
(5) In this section, evidence with respect to the human milk fortifier means
(a) evidence that establishes that the human milk fortifier is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use; and
(b) the results of tests carried out to determine the expiration date of the human milk fortifier.
B.25.019 (1) Even if the Minister has notified the manufacturer of a human milk fortifier that the sale or advertisement for sale of the human milk fortifier is authorized, it may only be sold in the following situations:
(a) it is sold by the manufacturer to
(i) a hospital, or
(ii) an individual, if
(A) the individual has received a written order for a human milk fortifier from a physician or from a nurse practitioner or dietitian who is authorized to write the order under the laws of a province, and
(B) the manufacturer has received a written request from a hospital that specifies the product name and quantity of the human milk fortifier that needs to be provided to the individual; or
(b) it is sold by a hospital to an individual who has received a written order for a human milk fortifier from a physician or from a nurse practitioner or dietitian who is authorized to write the order under the laws of a province.
(2) The following definitions apply in this section.
- dietitian
dietitian means a person who is registered and entitled under the laws of a province to practise as a dietitian and who is practising as a dietitian under those laws in that province. (diététiste)
- hospital
hospital has the same meaning as in section B.24.001. (hôpital)
- nurse practitioner
nurse practitioner has the same meaning as in section 1 of the New Classes of Practitioners Regulations. (infirmier praticien)
- physician
physician has the same meaning as in section B.24.001. (médecin)
B.25.020 (1) The following information is required to be displayed on the outer label of a human milk fortifier:
(a) a statement of the quantity, expressed in grams, of protein, fat, available carbohydrate and, where present, fibre in a quantity of human milk fortifier specified in the directions for use;
(b) a statement of the energy value, expressed in Calories, in a quantity of human milk fortifier specified in the directions for use;
(c) a statement of the quantity, expressed in milligrams, micrograms or International Units, of all vitamins, mineral nutrients and amino acids set out in item 6, Column II, of the table to section D.03.002 that are present in a quantity of human milk fortifier specified in the directions for use;
(d) a statement of the quantity, expressed in grams, milligrams, micrograms or International Units, of any other nutritive substance that is present in a quantity of human milk fortifier specified in the directions for use;
(e) directions for the storage of the human milk fortifier before and after the package has been opened;
(f) the directions for use for the human milk fortifier, including directions for the preparation, use and storage of the mixture of human milk fortifier and human milk;
(g) a statement indicating that the human milk fortifier is to be used only under medical supervision;
(h) the expiration date of the human milk fortifier; and
(i) the lot number of the human milk fortifier.
(2) If the human milk fortifier does not have an outer label,
(a) the information referred to in subsection (1) is required to be displayed on the inner label, except as otherwise provided in paragraph (b); and
(b) the information referred to in paragraphs (1)(e) and (f) may, despite section A.01.016, be displayed on a leaflet that is affixed or attached to the container that is in direct contact with the human milk fortifier.
(3) If the human milk fortifier has an outer label, the information referred to in paragraphs (1)(h) and (i) is also required to be displayed on the inner label.
B.25.021 It is prohibited, on the label of or in any advertisement for a human milk fortifier, to make any statement or claim relating to the content in the human milk fortifier of
(a) the percentage of the daily value of
(i) fat,
(ii) saturated fatty acids and trans fatty acids,
(iii) sodium,
(iv) potassium,
(v) sugars,
(vi) fibre, or
(vii) cholesterol; or
(b) the number of Calories from
(i) fat, or
(ii) saturated fatty acids and trans fatty acids.
Human Milk Substitutes and Food Containing Human Milk Substitutes
B.25.045 The common name of a human milk substitute or a new human milk substitute shall be infant formula. (préparation pour nourrissons)
- SOR/90-174, s. 2
B.25.046 (1) No person shall sell or advertise for sale a new human milk substitute unless the manufacturer, at least 90 days before the sale or advertisement, notifies the Minister in writing of the intention to sell or advertise for sale the new human milk substitute.
(2) The notification referred to in subsection (1) shall be signed and shall include, in respect of the new human milk substitute, the following information:
(a) the name under which it will be sold or advertised for sale;
(b) the name and the address of the principal place of business of the manufacturer;
(c) the names and addresses of each establishment in which it is manufactured;
(d) a list of all of its ingredients, stated quantitatively;
(e) the specifications for nutrient, microbiological and physical quality for the ingredients and for the new human milk substitute;
(f) details of quality control procedures respecting the testing of the ingredients and of the new human milk substitute;
(g) details of the manufacturing process and quality control procedures used throughout the process;
(h) the results of tests carried out to determine the expiration date of the new human milk substitute;
(i) the evidence relied on to establish that the new human milk substitute is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use;
(j) a description of the type of packaging to be used;
(k) directions for use;
(l) the written text of all labels, including package inserts, to be used in connection with the new human milk substitute; and
(m) the name and title of the person who signed the notification and the date of signature.
(3) Notwithstanding subsection (1), a person may sell or advertise for sale a new human milk substitute if the manufacturer has notified the Minister pursuant to subsection (1) and is informed in writing by the Minister that the notification is satisfactory.
- SOR/90-174, s. 2
- SOR/2018-69, ss. 27, 30(F)
B.25.047 Sections B.25.051 to B.25.059 apply in respect of new human milk substitutes.
- SOR/90-174, s. 2
- SOR/2003-11, s. 25
B.25.048 (1) No person shall sell or advertise for sale a human milk substitute that has undergone a major change unless the manufacturer of the human milk substitute, at least 90 days before the sale or advertisement, notifies the Minister in writing of the intention to sell or advertise for sale the human milk substitute.
(2) The notification referred to in subsection (1) shall be signed and shall include, in respect of the human milk substitute, the following information:
(a) the name under which it will be sold or advertised for sale;
(b) the name and the address of the principal place of business of the manufacturer;
(c) a description of the major change;
(d) the evidence relied on to establish that the human milk substitute is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use;
(e) the evidence relied on to establish that the major change has had no adverse effect on the human milk substitute;
(f) the written text of all labels, including package inserts, to be used in connection with the human milk substitute; and
(g) the name and title of the person who signed the notification and the date of signature.
(3) Notwithstanding subsection (1), a person may sell or advertise for sale a human milk substitute that has undergone a major change if the manufacturer has notified the Minister pursuant to subsection (1) and is informed in writing by the Minister that the notification is satisfactory.
- SOR/90-174, s. 2
- SOR/2018-69, ss. 27, 30(F)
B.25.050 [Repealed, SOR/90-174, s. 2]
B.25.051 (1) No person shall sell or advertise for sale a human milk substitute unless, when prepared according to directions for use, it complies with the provisions of this Division respecting human milk substitutes.
(2) No person shall sell or advertise for sale a food that is represented as containing a human milk substitute unless the human milk substitute portion of the food complies with the nutritional requirements set out in this Division for human milk substitutes.
- SOR/83-933, s. 1
B.25.052 (1) No person shall sell or advertise for sale a human milk substitute unless it meets the nutritional requirements of infants with normal or special dietary needs and it is of such a consistency that, when ready-to-serve, it passes freely through a nursing bottle nipple.
(2) No person shall sell or advertise for sale a food that is represented as containing a human milk substitute unless the human milk substitute portion of the food meets the nutritional requirements of infants with normal or special dietary needs.
- SOR/78-637, s. 6
- SOR/82-768, s. 76
- SOR/83-933, s. 1
B.25.053 (1) No person shall sell or advertise for sale a human milk substitute that requires, when prepared according to directions for use, the addition of a nutritive substance, other than water or a source of carbohydrate or both.
(2) No person shall sell or advertise for sale a food represented as containing a human milk substitute that requires, when prepared according to directions for use, the addition of a nutritive substance other than water.
- SOR/78-637, s. 7
- SOR/83-933, s. 1
B.25.054 (1) Except as otherwise provided in this Division, no person shall sell or advertise for sale a human milk substitute unless it contains, when prepared according to directions for use,
(a) per 100 available kilocalories
(i) not less than 3.3 and not more than 6.0 grams of fat,
(ii) not less than 500 milligrams of linoleic acid in the form of a glyceride,
(iii) not more than 1 kilocalorie from C22 Monoenoic Fatty Acids,
(iv) not less than 1.8 and not more than 4.0 grams of protein,
(v) not less than 1.8 grams of protein of nutritional quality equivalent to casein, or such an amount and quality of protein, including those proteins to which amino acids are added, that, when the quality of the protein is expressed as a fraction of the quality of casein,
(A) the fraction will not be less than 85/100, and
(B) the product obtained by multiplying the fraction by the gram weight of the protein will not be less than 1.8,
(vi) notwithstanding sections D.01.010, D.01.011 and D.02.009, the vitamin and mineral nutrient set out in Column I of an item of Table II to this Division in an amount not less than the amount set out in Column II of that item and not more than the amount set out in Column III of that item, and
(vii) not less than 12 milligrams of choline; and
(b) a ratio of
(i) alpha-tocopherol to linoleic acid of not less than 0.6 International Units to one gram,
(ii) calcium to phosphorus of not less than 1.2 grams to one gram and not more than 2.0 grams to one gram, and
(iii) vitamin B6 to protein of not less than 15 micrograms to one gram.
(2) No person shall sell or advertise for sale a food that is represented as containing a human milk substitute unless the human milk substitute portion of the food complies with subsection (1).
- SOR/78-637, s. 7
- SOR/83-933, s. 1
B.25.055 (1) Subparagraph B.25.054(1)(a)(i) does not apply to a human milk substitute represented as being for a fat-modified diet.
(2) Subparagraph B.25.054(1)(a)(iv), except that portion thereof that prescribes the maximum amount of protein, and subparagraph B.25.054(1)(a)(v) do not apply to a human milk substitute represented as being for a low (naming the amino acid) diet.
(3) All that portion of subparagraph B.25.054(1)(a)(vi) that prescribes the minimum amounts of vitamin D, calcium and phosphorus and subparagraph B.25.054(1)(b)(ii) do not apply to a human milk substitute represented as being for a low (naming the mineral) diet or a low vitamin D diet or both.
- SOR/83-933, s. 1
B.25.056 No person shall sell a human milk substitute or a food that is represented as containing a human milk substitute
(a) that contains an added nutritive substance that is
(i) normally contained in human milk, and
(ii) not referred to in paragraph B.25.054(1)(a)
unless the amount of that substance present per 100 available kilocalories of the human milk substitute or human milk substitute portion of the food, when prepared according to directions for use, is equal to the amount thereof present per 100 available kilocalories of human milk; or
(b) that contains added amino acids unless
(i) the amino acids are required to improve the quality of the protein in the human milk substitute or human milk substitute portion of the food and are present in an amount not exceeding the minimum required for that purpose, or
(ii) the protein content of the human milk substitute or human milk substitute portion of the food is supplied by isolated amino acids or by protein hydrolysate, or both
and only the L forms of the amino acids have been added.
- SOR/78-637, s. 8
- SOR/83-933, s. 1
B.25.057 (1) The label of a human milk substitute shall carry the following information:
(a) a statement of the content of protein, fat, available carbohydrate, ash and, where present, crude fibre in the food
(i) in grams per 100 grams or in grams per 100 millilitres of the human milk substitute as offered for sale, and
(ii) in grams in a stated quantity of the human milk substitute when ready-to-serve;
(b) a statement of the energy value expressed in
(i) calories per 100 grams or calories per 100 millilitres of the human milk substitute as offered for sale, and
(ii) calories in a stated quantity of the human milk substitute when ready-to-serve;
(c) a statement of the quantity of all vitamins and mineral nutrients listed in Table II to this Division
(i) in International Units or milligrams per 100 grams or in International Units or milligrams per 100 millilitres of the human milk substitute as offered for sale, and
(ii) in International Units or milligrams in a stated quantity of the human milk substitute when ready-to-serve;
(d) a statement of the quantity of choline and of any added nutritive substance normally contained in human milk and not referred to in paragraph B.25.054(1)(a)
(i) in milligrams or grams per 100 grams, or in milligrams or grams per 100 millilitres, of the human milk substitute as offered for sale, and
(ii) in milligrams or grams in a stated quantity of the human milk substitute when ready-to-serve;
(e) adequate directions for the preparation, use and storage of the human milk substitute after the container has been opened; and
(f) the expiration date of the human milk substitute.
(2) The label of a food that is represented as containing human milk substitute shall carry the following information:
(a) a statement on the principal display panel of the proportion of the human milk substitute contained in the food as offered for sale in close proximity to any claim regarding the presence of the human milk substitute and given equal prominence to such a claim;
(b) the common name of the human milk substitute in the list of ingredients to be followed by a statement of all the components contained in the human milk substitute;
(c) a statement of
(i) the content of protein, fat, available carbohydrate, ash and, where present, crude fibre contained in the human milk substitute portion of the food, expressed in grams per 100 grams or per 100 millilitres of the human milk substitute portion of the food as offered for sale,
(ii) the energy value of the human milk substitute portion of the food expressed in calories per 100 grams or in calories per 100 millilitres of the human milk substitute portion of the food as offered for sale,
(iii) the quantity of all the vitamins and mineral nutrients set out in Table II to this Division that are contained in the human milk substitute portion of the food in International Units or milligrams per 100 grams or in International Units or milligrams per 100 millilitres of the human milk substitute portion of the food as offered for sale, and
(iv) the quantity of choline and of any added nutritive substance normally contained in human milk and not referred to in paragraph B.25.054(1)(a) contained in the human milk substitute portion of the food in milligrams or grams per 100 grams or in milligrams or grams per 100 millilitres of the human milk substitute portion of the food as offered for sale;
(d) a statement of
(i) the content of protein, fat, available carbohydrate, ash and, where present, crude fibre in grams per 100 grams or per 100 millilitres of the food as offered for sale and in grams per stated quantity of ready-to-serve food,
(ii) the energy value expressed in calories per 100 grams or in calories per 100 millilitres of the food as offered for sale and in grams per a stated quantity of the food when ready-to-serve,
(iii) the quantity of all vitamins and mineral nutrients set out in Table II to this Division in International Units or milligrams per 100 grams or in International Units or milligrams per 100 millilitres of the food as offered for sale and in International Units or milligrams in a stated quantity of the food when ready-to-serve, and
(iv) the quantity of choline and of any added nutritive substance normally contained in human milk and not referred to in paragraph B.25.054(1)(a) in milligrams or grams per 100 grams or in milligrams or grams per 100 millilitres of the food as offered for sale and in milligrams or grams in a stated quantity of the food when ready-to-serve;
(e) adequate directions for the preparation, use and storage of the food after the container has been opened; and
(f) the expiration date of the food.
- SOR/83-933, s. 1
- SOR/88-559, s. 30
B.25.058 Notwithstanding section D.02.005, no person shall make any claim with respect to the iron content of a human milk substitute except as required by paragraph B.25.057(1)(c), unless the human milk substitute contains at least one milligram of iron per 100 available kilocalories.
- SOR/78-637, s. 9(F)
- SOR/83-933, s. 1
B.25.059 No person shall, on the label of or in any advertisement for a human milk substitute or a food represented as containing a human milk substitute, make any statement or claim relating to the content in the food of
(a) the percentage of the daily value of
(i) fat,
(ii) saturated fatty acids and trans fatty acids,
(iii) sodium,
(iv) potassium,
(v) sugars,
(vi) fibre, or
(vii) cholesterol; or
(b) the number of Calories from
(i) fat, or
(ii) saturated fatty acids and trans fatty acids.
- SOR/2003-11, s. 26
- SOR/2016-305, s. 60
B.25.060 (1) Where the manufacturer of a human milk substitute or of a food that is represented as containing a human milk substitute is requested in writing by the Minister to submit on or before a specified day evidence with respect to the human milk substitute, the manufacturer shall make no further sales of that human milk substitute or food that is represented as containing human milk substitute after that day unless he has submitted the evidence requested.
(2) If the Minister determines that the evidence submitted by a manufacturer under subsection (1) is not sufficient, he or she shall so notify the manufacturer in writing.
(3) Where, pursuant to subsection (2), a manufacturer is notified that the evidence with respect to the human milk substitute is not sufficient, he shall make no further sales of that human milk substitute or of that food that is represented as containing the human milk substitute unless he submits further evidence and is notified in writing by the Minister that the further evidence is sufficient.
(4) In this section, evidence with respect to the human milk substitute means
(a) evidence that establishes that the human milk substitute is nutritionally adequate to promote acceptable growth and development in infants when consumed in accordance with the directions for use; and
(b) the results of tests carried out to determine the expiration date of the human milk substitute.
- SOR/83-933, s. 1
- SOR/88-424, s. 2
- SOR/90-174, s. 3
- SOR/2018-69, ss. 12, 27
Additional Rules
- SOR/2021-57, s. 14
B.25.061 (1) Subject to subsection (2), no person shall include on the label of a food any representation respecting the consumption of the food by an infant who is less than six months of age.
(2) Subsection (1) does not apply in respect of a human milk fortifier, human milk substitute or new human milk substitute.
- SOR/83-933, s. 1
- SOR/90-174, s. 4
- SOR/2021-57, s. 15
B.25.062 (1) Subject to subsection (2), it is prohibited to sell an infant food if the food contains a food additive.
(2) Subsection (1) does not apply to
(a) bakery products that are labelled or advertised for consumption by infants;
(b) the following foods that are labelled or advertised for consumption by infants and that contain ascorbic acid:
(i) fruit purées, and
(ii) cereals containing banana;
(c) infant cereal products that contain lecithin;
(d) foods that are labelled or advertised for consumption by infants and that contain citric acid;
(e) infant formula that contains the food additives set out in Tables IV and X to section B.16.100 for use in infant formula;
(f) infant formula that contains ingredients manufactured with food additives set out in Table V to section B.16.100;
(g) infant formula that contains concentrated or dried whey products manufactured with liquid whey to which sodium hexametaphosphate has been added;
(h) infant cereal products that contain amylase in accordance with Table V to section B.16.100;
(i) infant formula that contains ascorbyl palmitate or tocopherols; or
(j) infant formula that contains oils to which ascorbyl palmitate or tocopherols have been added.
- SOR/83-933, s. 1
- SOR/90-24, s. 4
- SOR/91-149, s. 6
- SOR/97-559, s. 1
- SOR/2010-40, s. 3
- SOR/2010-41, s. 8
- SOR/2010-94, s. 6
- SOR/2010-141, s. 3
- SOR/2012-105, s. 2
- SOR/2021-57, s. 16
TABLE I
Sodium Content in Infant Foods
Column I Column II Food Total Sodium in Grams per 100 Grams of Food 1 Junior Desserts 0.10 2 Junior Meat, Junior Meat Dinners, Junior Dinners, Junior Breakfasts 0.25 3 Junior Vegetables, Junior Soups 0.20 4 Strained Desserts 0.05 5 Strained Meats, Strained Meat Dinners, Strained Dinners, Strained Breakfasts 0.15 6 Strained Vegetables, Strained Soups 0.10 - SOR/78-637, s. 10
- SOR/83-933, s. 1
TABLE II
Item No. Column I Column II Column III Vitamin or Mineral nutrient Minimum amount per 100 available kilocalories Maximum amount per 100 available kilocalories B.1 Biotin 2 µg — F.1 Folic acid 4 µg — N.1 Niacin 250 µg — P.1 d-pantothenic acid 300 µg — R.1 Riboflavin 60 µg — T.1 Thiamine 40 µg — T.2 Alpha-tocopherol 0.6 I.U. — V.1 Vitamin A 250 I.U. 500 I.U. V.2 Vitamin B6 35 µg — V.3 Vitamin B12 0.15 µg — V.4 Vitamin C 8 mg — V.5 Vitamin D 40 I.U. 80 I.U. V.6 Vitamin K1 8 µg — C.1 Calcium 50 mg — C.2 Chloride 55 mg 150 mg C.3 Copper 60 µg — L.1 Iodine 5 µg — L.2 Iron 0.15 mg — M.1 Magnesium 6 mg — M.2 Manganese 5 µg — P.2 Phosphorous 25 mg — P.3 Potassium 80 mg 200 mg S.1 Sodium 20 mg 60 mg Z.1 Zinc 0.5 mg — - SOR/83-933, s. 1
- SOR/98-458, s. 7(F)
- SOR/2022-197, s. 7
DIVISION 26Food Irradiation
Interpretation
- SOR/2017-16, s. 2(F)
B.26.001 In this Division, irradiation means treatment with ionizing radiation.
- SOR/89-175, s. 3
- SOR/2017-16, s. 3
Application
B.26.002 This Division does not apply to foods exposed to ionizing radiation from a measuring instrument used to determine weight, estimate bulk solids, measure the total solids in liquids or perform other inspection procedures.
- SOR/89-175, s. 3
General
B.26.003 (1) Subject to subsection (2), no person shall sell a food that has been irradiated.
(2) A food that is set out in column 1 of the table to this Division that has been irradiated may be sold if both of the following requirements are met:
(a) the ionizing radiation is of a type and from a source set out in column 2 for the purpose of irradiation set out in column 3; and
(b) the ionizing radiation that is absorbed by the food is either within the range set out in columns 4 and 5 or, if there is no minimum absorbed dose set out in column 4, is not more than the maximum absorbed dose set out in column 5.
- SOR/89-175, s. 3
- SOR/2017-16, s. 4
Records
B.26.004 (1) A manufacturer who sells a food that has been irradiated shall keep on their premises, for at least two years after the date of the irradiation, a record that contains all of the following information:
(a) the name of the food that was irradiated and the quantity and lot numbers of the food;
(b) the purpose of the irradiation;
(c) the date of the irradiation;
(d) the dose of ionizing radiation that was absorbed by the food;
(e) the type and source of the ionizing radiation;
(f) a statement that indicates whether the food was previously irradiated and, if it was previously irradiated, the information referred to in paragraphs (a) to (e) in respect of that previous irradiation.
(2) A person who imports a food for sale in Canada that has been irradiated shall keep on their premises, for at least two years after the date of importation, a record of the information required by subsection (1).
- SOR/89-175, s. 3
- SOR/2017-16, s. 5
Changes to the Table
B.26.005 A request that a food be added or a change made to the table to this Division shall be accompanied by a submission to the Minister containing the following information:
(a) the purpose and details of the proposed irradiation — including the type and source of the ionizing radiation — and the proposed number of treatments and the minimum and maximum absorbed doses of the ionizing radiation;
(b) data indicating that the minimum dose of ionizing radiation proposed to be used accomplishes the intended purpose of the irradiation and the maximum dose of ionizing radiation proposed does not exceed the amount required to accomplish the purpose of the irradiation;
(c) information on the nature of the dosimeter used, the frequency of the dosimetry on the food and data pertaining to the dosimetry and phantoms used to assure that the dosimetry readings reflect the dose absorbed by the food during irradiation;
(d) data indicating the effects, if any, on the nutritional quality of the food, raw and ready-to-serve, under the proposed conditions of irradiation and any other processes that are combined with the irradiation;
(e) data establishing that the irradiated food has not been significantly altered in chemical, physical or microbiological characteristics to render the food unfit for human consumption;
(f) where the Minister so requests, data establishing that the proposed irradiation is safe under the conditions proposed for the irradiation;
(g) the recommended conditions of storage and shipment of the irradiated food including the time, temperature and packaging and a comparison of the recommended conditions for the same food that has not been irradiated;
(h) details of any other processes to be applied to the food prior to or after the proposed irradiation; and
(i) such other data as the Minister may require to establish that consumers and purchasers of the irradiated food will not be deceived or misled as to the character, value, composition, merit or safety of the irradiated food.
TABLE
| Item | Column 1 | Column 2 | Column 3 | Column 4 | Column 5 |
|---|---|---|---|---|---|
| Food | Type and Source of Ionizing Radiation | Purpose of Irradiation | Minimum Absorbed Dose (kGy) | Maximum Absorbed Dose (kGy) | |
| 1 | Potatoes (Solanum tuberosum L.) | Gamma radiation from cobalt-60 | To inhibit sprouting during storage | 0.15 | |
| 2 | Onions (Allium cepa) | Gamma radiation from cobalt-60 | To inhibit sprouting during storage | 0.15 | |
| 3 | Wheat, flour, whole wheat flour (Triticum spp.) | Gamma radiation from cobalt-60 | To control insect infestation in stored food | 0.75 | |
| 4 | Whole or ground spices and dehydrated seasoning preparations |
|
|
| |
|
|
| |||
|
|
| |||
| 5 | Fresh raw ground beef |
|
|
|
|
|
|
|
| ||
|
|
|
| ||
| |||||
|
|
|
| ||
|
|
|
| ||
| 6 | Frozen raw ground beef |
|
|
|
|
|
|
|
| ||
|
|
|
| ||
| |||||
|
|
|
| ||
|
|
|
|
- SOR/89-175, s. 3
- SOR/98-458, s. 7(F)
- SOR/2017-16, ss. 6, 7
- SOR/2018-69, s. 27
DIVISION 27Low-Acid Foods Packaged in Hermetically Sealed Containers
B.27.001 In this Division,
- commercially sterile
commercially sterile means the condition obtained in a food that has been processed by the application of heat, alone or in combination with other treatments, to render the food free from viable forms of microorganisms, including spores, capable of growing in the food at temperatures at which the food is designed normally to be held during distribution and storage; (stérilité commerciale)
- hermetically sealed container
hermetically sealed container means a container designed and intended to be secure against the entry of microorganisms, including spores; (récipient hermétiquement fermé)
- low-acid food
low-acid food means a food, other than an alcoholic beverage, where any component of the food has a pH greater than 4.6 and a water activity greater than 0.85; (aliment peu acide)
- refrigeration
refrigeration means exposure to a temperature of 4°C or less but does not mean frozen; (réfrigéré)
- shipping container
shipping container means a receptacle, package or wrapper in which containers of food are placed for transportation; (contenant d’expédition)
- water activity
water activity means the ratio of the water vapour pressure of a food to the vapour pressure of pure water, at the same temperature and pressure. (activité de l’eau)
- SOR/89-309, s. 1
B.27.002 (1) No person shall sell a low-acid food packaged in a hermetically sealed container unless the food is commercially sterile.
(2) Subsection (1) does not apply in respect of a low-acid food packaged in a hermetically sealed container where
(a) the low-acid food is kept under refrigeration and the statement “Keep Refrigerated” and “Garder réfrigéré” is carried on the principal display panel of the label of its container, as well as on the label of its shipping container; or
(b) the low-acid food is kept frozen and the statement “Keep Frozen” and “Garder congelé” is carried on the principal display panel of the label of its container, as well as on the label of its shipping container.
(3) Subsection (1) does not apply in respect of tomatoes or tomato products packaged in hermetically sealed containers where the tomatoes or tomato products have a pH of 4.7 or less after heat processing.
- SOR/89-309, s. 1
- SOR/91-149, s. 7
- SOR/2018-108, s. 399
B.27.003 No person shall sell a low-acid food packaged in a hermetically sealed container where the container
(a) is swollen;
(b) is not properly sealed; or
(c) has any defect that may adversely affect its hermetic seal.
- SOR/89-309, s. 1
B.27.004 (1) Where, in the opinion of the Minister, the sale of a low-acid food packaged in a hermetically sealed container may contravene section B.27.002 or B.27.003, the Minister may, by notice in writing, request that the manufacturer or importer of the food submit, on or before the date specified in the notice, evidence that establishes that the processes used to manufacture, process and package the food rendered and maintained the food commercially sterile.
(2) Where a manufacturer or an importer receives a notice issued pursuant to subsection (1), the manufacturer or importer shall make no further sales of the food on or after the day specified in the notice until he has submitted the evidence requested in that notice.
(3) Where the Minister is of the opinion that the evidence submitted by a manufacturer or importer pursuant to subsection (1) is not sufficient, the Minister shall notify the manufacturer or importer in writing that the evidence is not sufficient.
(4) Where, pursuant to subsection (3), a manufacturer or importer is notified that the evidence he has submitted is not sufficient, the manufacturer or importer shall make no further sales of the food until he submits further evidence and is notified in writing by the Minister that the further evidence is sufficient.
- SOR/89-309, s. 1
- SOR/2018-69, s. 27
B.27.005 No person shall sell a commercially sterile low-acid food packaged in a hermetically sealed container unless
(a) the label or container of the food bears a code or lot number that identifies, in a legible and permanent manner,
(i) the establishment in which the product was rendered commercially sterile, and
(ii) the day, month and year on which the food was rendered commercially sterile; and
(b) the exact meaning of each item in any code or lot number referred to in paragraph (a) is available to an inspector at the establishment or, where the food is imported, from the importer.
- SOR/89-309, s. 1
DIVISION 28Novel Foods
Interpretation
B.28.001 The definitions in this section apply in this Division.
- genetically modify
genetically modify means to change the heritable traits of a plant, animal or microorganism by means of intentional manipulation. (modifier génétiquement)
- major change
major change means, in respect of a food, a change in the food that, based on the manufacturer’s experience or generally accepted nutritional or food science theory, places the modified food outside the accepted limits of natural variations for that food with regard to
(a) the composition, structure or nutritional quality of the food or its generally recognized physiological effects;
(b) the manner in which the food is metabolized in the body; or
(c) the microbiological safety, the chemical safety or the safe use of the food. (changement majeur)
- novel food
novel food means any of the following substances and foods, other than a supplemental ingredient or supplemented food:
(a) a substance, including a microorganism, that does not have a history of safe use as a food;
(b) a food that has been manufactured, prepared, preserved or packaged by a process that
(i) has not been previously applied to that food, and
(ii) causes the food to undergo a major change; and
(c) a food that is derived from a plant, animal or microorganism that has been genetically modified such that
(i) the plant, animal or microorganism exhibits characteristics that were not previously observed in that plant, animal or microorganism,
(ii) the plant, animal or microorganism no longer exhibits characteristics that were previously observed in that plant, animal or microorganism, or
(iii) one or more characteristics of the plant, animal or microorganism no longer fall within the anticipated range for that plant, animal or microorganism. (aliment nouveau)
- SOR/99-392, s. 1
- SOR/2022-169, s. 20
Pre-Market Notification
B.28.002 (1) No person shall sell or advertise for sale a novel food unless the manufacturer or importer of the novel food
(a) has notified the Minister in writing of their intention to sell or advertise for sale the novel food; and
(b) has received a written notice from the Minister under paragraph B.28.003(1)(a) or subsection B.28.003(2).
(2) A notification referred to in paragraph (1)(a) shall be signed by the manufacturer or importer, or a person authorized to sign on behalf of the manufacturer or importer, and shall include the following information:
(a) the common name under which the novel food will be sold;
(b) the name and address of the principal place of business of the manufacturer and, if the address is outside Canada, the name and address of the principal place of business of the importer;
(c) a description of the novel food, together with
(i) information respecting its development,
(ii) details of the method by which it is manufactured, prepared, preserved, packaged and stored,
(iii) details of the major change, if any,
(iv) information respecting its intended use and directions for its preparation,
(v) information respecting its history of use as a food in a country other than Canada, if applicable, and
(vi) information relied on to establish that the novel food is safe for consumption;
(d) information respecting the estimated levels of consumption by consumers of the novel food;
(e) the text of all labels to be used in connection with the novel food; and
(f) the name and title of the person who signed the notification and the date of signing.
- SOR/99-392, s. 1
- SOR/2018-69, s. 27
B.28.003 (1) Within 45 days after receiving a notification referred to in paragraph B.28.002(1)(a), the Minister shall review the information included in the notification and
(a) if the information establishes that the novel food is safe for consumption, notify the manufacturer or importer in writing that the information is sufficient; or
(b) if additional information of a scientific nature is necessary in order to assess the safety of the novel food, request in writing that the manufacturer or importer submit that information.
(2) Within 90 days after receiving the additional information requested under paragraph (1)(b) the Minister shall assess it and, if it establishes that the novel food is safe for consumption, notify the manufacturer or importer in writing that the information is sufficient.
- SOR/99-392, s. 1
- SOR/2018-69, s. 27
DIVISION 29Supplemented Foods
Interpretation
B.29.001 (1) The following definitions apply in this Division.
- Directory of SFFT Formats
Directory of SFFT Formats means the document entitled Directory of Supplemented Food Facts Table Formats, published by the Government of Canada on its website, as amended from time to time. (Répertoire des modèles de TRAS)
- Directory of Supplemented Food Caution Identifier Specifications
Directory of Supplemented Food Caution Identifier Specifications means the document entitled Directory of Supplemented Food Caution Identifier Specifications, published by the Government of Canada on its website, as amended from time to time. (Répertoire des spécifications sur l’identifiant des aliments supplémentés avec mise en garde)
- fat
fat means all fatty acids expressed as triglycerides. (lipides)
(2) For the purposes of this Division and subject to subsection (3), the amount of vitamins must be determined in accordance with section D.01.003.
(3) For the purposes of this Division, the amount in metric units of the following vitamins must be determined in terms of their amount in the supplemented food in accordance with column 5 of the List of Permitted Supplemental Ingredients, as applicable, and expressed in the applicable unit set out in column 3 of the List of Permitted Supplemental Ingredients:
(a) beta-carotene as a form of vitamin A or retinol, including its derivatives, as a form of vitamin A, or both, if either is a supplemental ingredient; and
(b) niacin, if it is a supplemental ingredient.
Nutrition Labelling
Core Information
B.29.002 (1) Except as otherwise provided in this section and sections B.29.003 to B.29.005, B.29.018 and B.29.019, the label of a supplemented food must carry a supplemented food facts table that contains only the information set out in column 1 of the table to this section, expressed using a description set out in column 2, in the unit set out in column 3 and in the manner set out in column 4.
(2) For the purposes of subsection (1), the serving of stated size set out in the supplemented food facts table, as expressed in a metric unit, must be used as the basis for determining the information appearing in the supplemented food facts table in respect of the energy value of, and the content of nutrients and supplemental ingredients in, the supplemented food.
(3) Subject to subsection (8), the percentage of the daily value for a vitamin or mineral nutrient shown in the supplemented food facts table in accordance with subsection (1) must be established on the basis of the amount, by weight, of the vitamin or mineral nutrient per serving of stated size of the supplemented food, rounded off in the applicable manner set out in column 4 of the table to this section.
(4) If the information in respect of six or more of the energy value and nutrients referred to in column 1 of items 2 to 5 and 7 to 15 of the table to this section may be expressed as “0” in the supplemented food facts table in accordance with this section, the supplemented food facts table need only include the following information:
(a) the serving of stated size;
(b) the energy value;
(c) the amount of fat;
(d) the amount of carbohydrate;
(e) the amount of protein;
(f) the amount of any nutrient that is the subject of a representation on the label of the supplemented food, or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food, if the representation expressly or implicitly indicates that the supplemented food has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004;
(g) the amount of any added sugar alcohol;
(h) the amount of any supplemental ingredient;
(i) the amount of any vitamin or mineral nutrient that is declared as a component of an ingredient, other than flour, of the supplemented food;
(j) the amount of any nutrient referred to in column 1 of any of items 4, 5, 7, 8, 10, 11 and 13 to 15 of the table to this section that may not be expressed as “0” in the supplemented food facts table;
(k) the statement “Not a significant source of (naming each nutrient that is omitted from the supplemented food facts table in accordance with this subsection)” or, if the supplemented food meets the condition specified in subsection B.29.010(3), the statement “Not a significant source of other nutrients”;
(l) the % Daily Value interpretative statement; and
(m) the “Supplemented with” interpretative statement.
(5) The supplemented food facts table of a supplemented food that is a single-serving prepackaged product need only include the following information:
(a) the serving of stated size;
(b) the energy value;
(c) the amount of fat;
(d) the amount of carbohydrate;
(e) the amount of protein;
(f) the amount of any nutrient that is the subject of a representation on the label of the supplemented food, or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food, if the representation expressly or implicitly indicates that the supplemented food has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004;
(g) the amount of any added sugar alcohol;
(h) the amount of any supplemental ingredient;
(i) the amounts of saturated fatty acids and trans fatty acids and the sum of saturated fatty acids and trans fatty acids, if any of the amounts or the sum may not be expressed as “0” in the supplemented food facts table;
(j) the amount of any nutrient referred to in column 1 of item 8 or 11 of the table to this section that may not be expressed as “0” in the supplemented food facts table;
(k) the % Daily Value interpretative statement; and
(l) the “Supplemented with” interpretative statement.
(6) Subsection (1) does not apply to a supplemented food intended solely for use as an ingredient in the manufacture of other supplemented foods intended for sale to a consumer at the retail level.
(7) If the supplemented food facts table on the label of a supplemented food corresponds to Figure 6.5(B), 6.6(B), 6.5.1(B), 6.6.1(B), 7.3(B), 7.4(B), 7.3.1(B), 7.4.1(B), 17.2(E) and (F) or 17.2.1(E) and (F) of the Directory of SFFT Formats, the supplemented food facts table is not required to show the % Daily Value interpretative statement or the “Supplemented with” interpretative statement.
(8) Subject to subsection (10), if a substance has been added as a supplemental ingredient, the amount referred to in item 18 of the table to this section includes the total amount of the substance in the supplemented food, unless otherwise provided in column 3 or 5 of the List of Permitted Supplemental Ingredients.
(9) If any amount of a nutrient referred to in column 1 of the table to this section has been added as a supplemental ingredient, the amount of the nutrient may only be expressed in the supplemented food facts table in accordance with item 18 of the table to this section.
(10) If a substance other than a nutrient has been added as a supplemental ingredient and the substance has one or more constituents for which a maximum amount is specified in column 3 of the List of Permitted Supplemental Ingredients, the amount of each constituent set out in the supplemented food facts table must include the total amount of the constituent in the supplemented food, unless otherwise provided in column 3 or 5 of the List of Permitted Supplemental Ingredients.
TABLE
Core Information
Item Column 1 Column 2 Column 3 Column 4 Information Description Unit Manner of expression 1 Serving of stated size “Serving Size (naming the serving size)”, “Serving (naming the serving size)” or “Per (naming the serving size)” The size is expressed
(a) in the case of a supplemented food that is a single-serving prepackaged product,
(i) per package, and
(ii) in grams or millilitres, in accordance with subparagraph B.01.002A(2)(a)(i) or (ii); and
(b) in the case of a supplemented food that is a multiple-serving prepackaged product, in the following units set out in column 3B of the Table of Reference Amounts:
(i) the household measure that applies to the supplemented food, and
(ii) the metric measure that applies to the supplemented food.
(1) The size if expressed in a metric unit is rounded off
(a) if it is less than 10 g or 10 mL, to the nearest multiple of 0.1 g or 0.1 mL; and
(b) if it is 10 g or more or 10 mL or more, to the nearest multiple of 1 g or 1 mL.
(2) The size if expressed as a fraction is represented by a numerator and a denominator separated by a line.
(3) The size must include the word “assorted” if the information in the supplemented food facts table of a prepackaged product containing an assortment of supplemented foods is set out as a composite value.
2 Energy value “Calories”, “Total Calories” or “Calories, Total” The value is expressed in Calories per serving of stated size. The value is rounded off
(a) if it is less than 5 Calories,
(i) if the supplemented food meets the conditions set out in column 2 of item 1 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of energy” set out in column 1, to “0” Calorie, and
(ii) in all other cases, to the nearest multiple of 1 Calorie;
(b) if it is 5 Calories or more but not more than 50 Calories, to the nearest multiple of 5 Calories; and
(c) if it is more than 50 Calories, to the nearest multiple of 10 Calories.
3 Amount of fat “Fat”, “Total Fat” or “Fat, Total” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g,
(i) if the supplemented food meets the conditions set out in column 2 of item 11 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of fat” set out in column 1 and the amounts of saturated fatty acids and trans fatty acids are declared as “0 g” in the supplemented food facts table or are omitted from that table in accordance with subsection B.29.002(4) and no other fatty acids are declared in an amount greater than 0 g, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
4 Amount of saturated fatty acids “Saturated Fat”, “Saturated Fatty Acids”, “Saturated” or “Saturates” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g,
(i) if the supplemented food meets the conditions set out in column 2 of item 18 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of saturated fatty acids” set out in column 1, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
5 Amount of trans fatty acids “Trans Fat”, “Trans Fatty Acids” or “Trans” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g,
(i) if the supplemented food meets the conditions set out in column 2 of item 22 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of trans fatty acids” set out in column 1, to “0 g”, and
(ii) in all other cases, to the nearest multiple of 0.1 g;
(b) if it is 0.5 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
6 The sum of saturated fatty acids and trans fatty acids “Saturated Fat + Trans Fat”, “Saturated Fatty Acids + Trans Fatty Acids”, “Saturated + Trans” or “Saturates + Trans” The sum is expressed as a percentage of the daily value per serving of stated size. The percentage is rounded off
(a) if the amounts of saturated fatty acids and trans fatty acids are declared as “0 g”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
7 Amount of cholesterol “Cholesterol” The amount
(a) is expressed in milligrams per serving of stated size; and
(b) may also be expressed as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if the supplemented food meets the conditions set out in column 2 of item 27 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of cholesterol” set out in column 1, to “0 mg”; and
(b) in all other cases, to the nearest multiple of 5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
8 Amount of sodium “Sodium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg,
(i) if the supplemented food meets the conditions set out in column 2 of item 31 of the Table of Permitted Nutrient Content Statements and Claims for the subject “free of sodium or salt” set out in column 1, to “0 mg”, and
(ii) in all other cases, to the nearest multiple of 1 mg;
(b) if it is 5 mg or more but not more than 140 mg, to the nearest multiple of 5 mg; and
(c) if it is more than 140 mg, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
9 Amount of carbohydrate “Carbohydrate”, “Total Carbohydrate” or “Carbohydrate, Total” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
10 Amount of fibre “Fibre”, “Fiber”, “Dietary Fibre” or “Dietary Fiber” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
11 Amount of sugars “Sugars” The amount is expressed
(a) in grams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
(2) The percentage is rounded off
(a) if the amount is declared as “0 g”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
12 Amount of protein “Protein” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to the nearest multiple of 0.1 g; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
13 Amount of potassium “Potassium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
14 Amount of calcium “Calcium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
15 Amount of iron “Iron” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
16 % Daily Value interpretative statement “*5% or less is a little, 15% or more is a lot” [not applicable] The “% Daily Value” or “% DV” subheading is followed by an asterisk in order to reference the % Daily Value interpretative statement shown in the supplemented food facts table. 17 “Supplemented with” interpretative statement “† Includes naturally occurring and supplemental amounts” [not applicable] The “Supplemented with” subheading is followed by a dagger in order to reference the “Supplemented with” interpretative statement shown in the supplemented food facts table. 18 Amount of supplemental ingredient The supplemental ingredient is described in accordance with column 1 of the List of Permitted Supplemental Ingredients. The amount is expressed in
(a) the applicable unit referred to in column 3 of the List of Permitted Supplemental Ingredients, per serving of stated size; and
(b) in the case of a nutrient with a daily value, as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off to the nearest whole number and expressed in the manner set out in column 3 and, if applicable, column 5 of the List of Permitted Supplemental Ingredients.
(2) Unless otherwise provided in column 5 of the List of Permitted Supplemental Ingredients, the percentage is rounded off
(a) if the amount declared in the applicable unit referred to in column 3 of the List of Permitted Supplemental Ingredients is “0”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
Additional Information
B.29.003 (1) Subject to subsection (2), the supplemented food facts table may also contain information set out in column 1 of the table to this section.
(2) This section does not apply in respect of a vitamin or mineral nutrient that is set out in the supplemented food facts table in accordance with subsection B.29.002(1) if any amount of that vitamin or mineral nutrient has been added as a supplemental ingredient.
(3) If information set out in column 1 of the table to this section is included in the supplemented food facts table, it must be expressed using a description set out in column 2, in the unit set out in column 3 and in the manner set out in column 4.
(4) For the purposes of subsection (3), the serving of stated size set out in the supplemented food facts table, as expressed in a metric unit, must be used as the basis for determining the information appearing in the supplemented food facts table in respect of the energy value and nutrient content of the supplemented food.
(5) The percentage of the daily value for a vitamin or mineral nutrient shown in the supplemented food facts table in accordance with subsection (3) must be established on the basis of the amount, by weight, of the vitamin or mineral nutrient per serving of stated size for the supplemented food, rounded off in the applicable manner set out in column 4 of the table to this section.
(6) The amount of omega-6 polyunsaturated fatty acids, omega-3 polyunsaturated fatty acids and monounsaturated fatty acids must be shown in the supplemented food facts table if
(a) the amount of any of those groups of fatty acids or the amount of polyunsaturated fatty acids is shown in the supplemented food facts table or on the label of the supplemented food or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food; or
(b) the amount of any specific fatty acid is shown on the label of the supplemented food or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food.
(7) If the label of the supplemented food, or any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer, contains a representation, express or implied, that includes information that is set out in column 1 of the table to this section, that information must also be shown in the supplemented food facts table.
(8) The supplemented food facts table must show the amount of any added sugar alcohol.
(9) The supplemented food facts table must show the amount of any vitamin or mineral nutrient that is declared as a component of an ingredient, other than flour.
(10) If information set out in column 1 of the table to this section is included in the supplemented food facts table, it must be shown
(a) in English and French; or
(b) in one of those languages, if, in accordance with subsection B.01.012(3), the information that is required by these Regulations to be shown on the label may be shown in that language only and is shown on the label in that language.
TABLE
Additional Information
Item Column 1 Column 2 Column 3 Column 4 Information Description Unit Manner of expression 1 Servings per package “Servings per Container”, “(number of units) per Container”, “Servings per Package”, “(number of units) per Package”, “Servings per (naming the package type)”, or “(number of units) per (naming the package type)” The quantity is expressed in number of servings. (1) The quantity is rounded off
(a) if it is less than 2, to the nearest multiple of 1;
(b) if it is between 2 and 5, to the nearest multiple of 0.5; and
(c) if it is more than 5, to the nearest multiple of 1.
(2) If a quantity is rounded off, it must be preceded by the word “about”.
(3) If the product is of a random weight, the quantity may be declared as “varied”.
2 Energy value “kilojoules” or “kJ” The value is expressed in kilojoules per serving of stated size. The value is rounded off to the nearest multiple of 10 kilojoules. 3 Amount of polyunsaturated fatty acids “Polyunsaturated Fat”, “Polyunsaturated Fatty Acids”, “Polyunsaturated” or “Polyunsaturates” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
4 Amount of omega-6 polyunsaturated fatty acids (1) If the supplemented food facts table includes the amount of polyunsaturated fatty acids: “Omega-6”, “Omega-6 Polyunsaturated Fat”, “Omega-6 Polyunsaturated Fatty Acids”, “Omega-6 Polyunsaturates” or “Omega-6 Polyunsaturated”
(2) In all other cases: “Omega-6 Polyunsaturated Fat”, “Omega-6 Polyunsaturated Fatty Acids”, “Omega-6 Polyunsaturates” or “Omega-6 Polyunsaturated”
The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
5 Amount of omega-3 polyunsaturated fatty acids (1) If the supplemented food facts table includes the amount of polyunsaturated fatty acids: “Omega-3”, “Omega-3 Polyunsaturated Fat”, “Omega-3 Polyunsaturated Fatty Acids”, “Omega-3 Polyunsaturates” or “Omega-3 Polyunsaturated”
(2) In all other cases: “Omega-3 Polyunsaturated Fat”, “Omega-3 Polyunsaturated Fatty Acids”, “Omega-3 Polyunsaturates” or “Omega-3 Polyunsaturated”
The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
6 Amount of monounsaturated fatty acids “Monounsaturated Fat”, “Monounsaturated Fatty Acids”, “Monounsaturates” or “Monounsaturated” The amount is expressed in grams per serving of stated size. The amount is rounded off
(a) if it is less than 1 g, to the nearest multiple of 0.1 g;
(b) if it is 1 g or more but not more than 5 g, to the nearest multiple of 0.5 g; and
(c) if it is more than 5 g, to the nearest multiple of 1 g.
7 Amount of soluble fibre “Soluble Fibre” or “Soluble Fiber” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
8 Amount of insoluble fibre “Insoluble Fibre” or “Insoluble Fiber” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
9 Amount of sugar alcohol (1) If the supplemented food contains only one type of sugar alcohol: “Sugar Alcohol”, “Polyol” or “(naming the sugar alcohol)”
(2) In all other cases: “Sugar Alcohols” or “Polyols”
The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
10 Amount of starch “Starch” The amount is expressed as grams per serving of stated size. The amount is rounded off
(a) if it is less than 0.5 g, to “0 g”; and
(b) if it is 0.5 g or more, to the nearest multiple of 1 g.
11 Amount of vitamin A “Vitamin A” or “Vit A” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 µg, to “0 µg”;
(b) if it is 5 µg or more but less than 50 µg, to the nearest multiple of 10 µg;
(c) if it is 50 µg or more but less than 250 µg, to the nearest multiple of 50 µg; and
(d) if it is 250 µg or more, to the nearest multiple of 100 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
12 Amount of vitamin C “Vitamin C” or “Vit C” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 mg, to “0 mg”;
(b) if it is 0.1 mg or more but less than 1 mg, to the nearest multiple of 0.2 mg;
(c) if it is 1 mg or more but less than 5 mg, to the nearest multiple of 0.5 mg; and
(d) if it is 5 mg or more, to the nearest multiple of 1 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
13 Amount of vitamin D “Vitamin D” or “Vit D” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 µg, to “0 µg”;
(b) if it is 0.1 µg or more but less than 1 µg, to the nearest multiple of 0.2 µg;
(c) if it is 1 µg or more but less than 5 µg, to the nearest multiple of 0.5 µg; and
(d) if it is 5 µg or more, to the nearest multiple of 1 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
14 Amount of vitamin E “Vitamin E” or “Vit E” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
15 Amount of vitamin K “Vitamin K” or “Vit K” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
16 Amount of thiamine “Thiamine”, “Thiamin”, “Thiamine (Vitamin B1)”, “Thiamine (Vit B1)”, “Thiamin (Vitamin B1)” or “Thiamin (Vit B1)” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
17 Amount of riboflavin “Riboflavin”, “Riboflavin (Vitamin B2)” or “Riboflavin (Vit B2)” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
18 Amount of niacin “Niacin” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
19 Amount of vitamin B6 “Vitamin B6” or “Vit B6” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
20 Amount of folate “Folate” The amount is expressed
(a) in micrograms of dietary folate equivalents (DFE) per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 µg DFE, to “0 µg DFE”;
(b) if it is 1 µg DFE or more but less than 10 µg DFE, to the nearest multiple of 2 µg DFE;
(c) if it is 10 µg DFE or more but less than 50 µg DFE, to the nearest multiple of 5 µg DFE; and
(d) if it is 50 µg DFE or more, to the nearest multiple of 10 µg DFE.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg DFE”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
21 Amount of vitamin B12 “Vitamin B12” or “Vit B12” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 µg, to “0 µg”;
(b) if it is 0.005 µg or more but less than 0.05 µg, to the nearest multiple of 0.01 µg;
(c) if it is 0.05 µg or more but less than 0.25 µg, to the nearest multiple of 0.025 µg; and
(d) if it is 0.25 µg or more, to the nearest multiple of 0.05 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
22 Amount of biotin “Biotin” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
23 Amount of pantothenic acid “Pantothenic Acid” or “Pantothenate” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.01 mg, to “0 mg”;
(b) if it is 0.01 mg or more but less than 0.1 mg, to the nearest multiple of 0.02 mg;
(c) if it is 0.1 mg or more but less than 0.5 mg, to the nearest multiple of 0.05 mg; and
(d) if it is 0.5 mg or more, to the nearest multiple of 0.1 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
24 Amount of choline “Choline” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 mg, to “0 mg”;
(b) if it is 1 mg or more but less than 10 mg, to the nearest multiple of 2 mg;
(c) if it is 10 mg or more but less than 50 mg, to the nearest multiple of 5 mg; and
(d) if it is 50 mg or more, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
25 Amount of phosphorous “Phosphorus” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
26 Amount of iodide “Iodide” or “Iodine” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 µg, to “0 µg”;
(b) if it is 1 µg or more but less than 10 µg, to the nearest multiple of 2 µg;
(c) if it is 10 µg or more but less than 50 µg, to the nearest multiple of 5 µg; and
(d) if it is 50 µg or more, to the nearest multiple of 10 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
27 Amount of magnesium “Magnesium” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 1 mg, to “0 mg”;
(b) if it is 1 mg or more but less than 10 mg, to the nearest multiple of 2 mg;
(c) if it is 10 mg or more but less than 50 mg, to the nearest multiple of 5 mg; and
(d) if it is 50 mg or more, to the nearest multiple of 10 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
28 Amount of zinc “Zinc” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 mg, to “0 mg”;
(b) if it is 0.05 mg or more but less than 0.5 mg, to the nearest multiple of 0.1 mg;
(c) if it is 0.5 mg or more but less than 2.5 mg, to the nearest multiple of 0.25 mg; and
(d) if it is 2.5 mg or more, to the nearest multiple of 0.5 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
29 Amount of selenium “Selenium” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.1 µg, to “0 µg”;
(b) if it is 0.1 µg or more but less than 1 µg, to the nearest multiple of 0.2 µg;
(c) if it is 1 µg or more but less than 5 µg, to the nearest multiple of 0.5 µg; and
(d) if it is 5 µg or more, to the nearest multiple of 1 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
30 Amount of copper “Copper” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.0015 mg, to “0 mg”;
(b) if it is 0.0015 mg or more but less than 0.025 mg, to the nearest multiple of 0.002 mg;
(c) if it is 0.025 mg or more but less than 0.05 mg, to the nearest multiple of 0.005 mg; and
(d) if it is 0.05 mg or more, to the nearest multiple of 0.01 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
31 Amount of manganese “Manganese” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.005 mg, to “0 mg”;
(b) if it is 0.005 mg or more but less than 0.05 mg, to the nearest multiple of 0.01 mg;
(c) if it is 0.05 mg or more but less than 0.25 mg, to the nearest multiple of 0.025 mg; and
(d) if it is 0.25 mg or more, to the nearest multiple of 0.05 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
32 Amount of chromium “Chromium” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
33 Amount of molybdenum “Molybdenum” The amount is expressed
(a) in micrograms per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 0.05 µg, to “0 µg”;
(b) if it is 0.05 µg or more but less than 0.5 µg, to the nearest multiple of 0.1 µg;
(c) if it is 0.5 µg or more but less than 2.5 µg, to the nearest multiple of 0.25 µg; and
(d) if it is 2.5 µg or more, to the nearest multiple of 0.5 µg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 µg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
34 Amount of chloride “Chloride” The amount is expressed
(a) in milligrams per serving of stated size; and
(b) as a percentage of the daily value per serving of stated size.
(1) The amount is rounded off
(a) if it is less than 5 mg, to “0 mg”;
(b) if it is 5 mg or more but less than 50 mg, to the nearest multiple of 10 mg;
(c) if it is 50 mg or more but less than 250 mg, to the nearest multiple of 25 mg; and
(d) if it is 250 mg or more, to the nearest multiple of 50 mg.
(2) The percentage is rounded off
(a) if the amount is declared as “0 mg”, to 0%; and
(b) in all other cases, to the nearest multiple of 1%.
Supplemented Foods for Use in Manufacturing Other Supplemented Foods
B.29.004 (1) This section applies to a supplemented food intended solely for use as an ingredient in the manufacture of other supplemented foods intended for sale to a consumer at the retail level.
(2) It is prohibited to sell the supplemented food unless the information referred to in subsection (3) in respect of the supplemented food is provided in writing and accompanies the supplemented food when it is delivered to the purchaser.
(3) The information
(a) must include the information that would, but for subsection B.29.002(6), be required by sections B.29.002 and B.29.003 to be included in a supplemented food facts table;
(b) may include other information that is permitted by section B.29.003 to be included in that supplemented food facts table; and
(c) must be expressed in accordance with sections B.29.002 and B.29.003, subject to the following modifications, namely,
(i) information in respect of supplemental ingredients must be expressed according to the applicable unit referred to in column 3 of the List of Permitted Supplemental Ingredients,
(A) per gram or 100 g of the supplemented food, if the net quantity of the supplemented food is declared on the label by weight or by count, or
(B) per millilitre or 100 mL of the supplemented food, if the net quantity of the supplemented food is declared on the label by volume,
(ii) information — other than in respect of supplemental ingredients — for vitamins referred to in subsection D.01.002(1) must be expressed in the applicable unit referred to in subsection D.01.003(1) and for mineral nutrients referred to in paragraphs D.02.001(1)(a) to (j), (l) to (n) and (p) must be expressed in milligrams for sodium, potassium, calcium, phosphorus, magnesium, iron, zinc, chloride, copper and manganese and in micrograms for iodide, chromium, selenium and molybdenum,
(A) per gram or 100 g of the supplemented food, if the net quantity of the supplemented food is declared on the label by weight or by count, or
(B) per millilitre or 100 mL of the supplemented food, if the net quantity of the supplemented food is declared on the label by volume,
(iii) information for other nutrients and the energy value set out in column 1 of the table to section B.29.002 or the table to section B.29.003 must be expressed in the units referred to in column 3,
(A) per gram or 100 g of the supplemented food, if the net quantity of the supplemented food is declared on the label by weight or by count, or
(B) per millilitre or 100 mL of the supplemented food, if the net quantity of the supplemented food is declared on the label by volume,
(iv) percentages of daily values and information on servings of stated size may be omitted, and
(v) the information must be stated with a degree of precision that corresponds to the accuracy of the analytical methodology used to produce the information.
Basis of Information
B.29.005 (1) Subject to subsections (2) to (5), the information in the supplemented food facts table must be set out only on the basis of the supplemented food as offered for sale.
(2) If a prepackaged product contains an assortment of supplemented foods of the same type and the typical serving consists of only one of those supplemented foods, the information in the supplemented food facts table must be set out
(a) on the basis of each of the supplemented foods contained in the prepackaged product, if the information set out in column 1 of the table to section B.29.002 for each of those supplemented foods is different; or
(b) on the basis of one of the supplemented foods contained in the prepackaged product, if the information set out in column 1 of the table to section B.29.002 for each of those supplemented foods is the same.
(3) If a prepackaged product contains an assortment of supplemented foods of the same type and the typical serving consists of more than one of those supplemented foods, the information in the supplemented food facts table must be set out for each of the supplemented foods contained in the prepackaged product or as a composite value.
(4) If a supplemented food is to be prepared in accordance with directions provided in or on the package or is commonly combined with other ingredients or another food or cooked before being consumed, the supplemented food facts table may also set out information for the supplemented food as prepared, in which case
(a) the supplemented food facts table must set out the following information for the supplemented food as prepared, namely,
(i) except in the case described in subparagraph (ii), the amount of the supplemented food expressed using the unit referred to in column 3 of subparagraph 1(b)(i) of the table to section B.29.002 as “about (naming the serving size)” or “about (naming the serving size) prepared” and, if applicable, in the manner specified in column 4 of subitems 1(1) and (2),
(ii) if the supplemented food is commonly served combined with another food, the amount of the other food expressed using the unit referred to in column 3 of subparagraph 1(b)(i) of the table to section B.29.002,
(iii) the energy value, expressed using a description set out in column 2 of item 2 of the table to section B.29.002, in the unit set out in column 3 and in the manner set out in column 4,
(iv) the information set out in column 1 of items 3, 6 to 8, 10, 11 and 13 to 15 of the table to section B.29.002 and in column 1 of items 11 to 34 of the table to section B.29.003 that is declared as a percentage of the daily value in the supplemented food facts table for the supplemented food as sold, expressed using a description set out in column 2 of those tables, as a percentage of the daily value per serving of stated size and in the manner specified in column 4 of those tables, and
(v) the information referred to in column 1 of item 18 of the table to section B.29.002, expressed using the description referred to in column 2, in the unit referred to in column 3 and in the manner referred to in column 4; and
(b) the supplemented food facts table may also set out the following information for the added ingredients or the other food, if it is declared in the supplemented food facts table for the supplemented food as sold, namely,
(i) the information set out in column 1 of items 3 to 5 and 7 to 12 of the table to section B.29.002, expressed using a description set out in column 2, in milligrams for the information set out in column 1 of items 7 and 8 and in grams for the information set out in column 1 of items 3 to 5 and 9 to 12 and in the manner specified in column 4,
(ii) the information set out in column 1 of items 3 to 10 of the table to section B.29.003, expressed using a description set out in column 2, in grams and in the manner specified in column 4, and
(iii) the information set out in column 1 of item 2 of the table to section B.29.002, expressed using a description set out in column 2, in the unit set out in column 3 per serving of stated size of the supplemented food as prepared, and in the manner specified in column 4.
(5) The information in the supplemented food facts table may also be set out on the basis of other amounts of the supplemented food that reflect different uses or different units of measurement of the supplemented food, in which case
(a) the supplemented food facts table must set out the following information for each of the other amounts of the supplemented food, namely,
(i) the amount of the supplemented food expressed in a household measure and a metric measure and in the manner specified in column 4 of subitems 1(1) and (2) of the table to section B.29.002,
(ii) the energy value, expressed using a description set out in column 2 of item 2 of the table to section B.29.002, in the unit set out in column 3 and in the manner set out in column 4,
(iii) the information set out in column 1 of items 3, 6 to 8, 10, 11 and 13 to 15 of the table to section B.29.002 and in column 1 of items 11 to 34 of the table to section B.29.003 that is declared as a percentage of the daily value in the supplemented food facts table for the first amount of the supplemented food for which information is declared, expressed using a description set out in column 2 of those tables, as a percentage of the daily value per serving of stated size and in the manner specified in column 4 of those tables, and
(iv) the information referred to in column 1 of item 18 of the table to section B.29.002, expressed using the description referred to in column 2, the unit referred to in column 3 and in the manner referred to in column 4; and
(b) if the supplemented food facts table is set out in a version of the aggregate format specified in section B.29.015, it must also set out the following information for each of the other amounts of the supplemented food, if that information is declared in the supplemented food facts table for the first amount of the supplemented food for which information is declared, namely,
(i) the information set out in column 1 of items 3 to 5 and 7 to 15 of the table to section B.29.002, expressed using a description set out in column 2, in milligrams for the information set out in column 1 of items 7, 8 and 13 to 15, and in grams for the information set out in column 1 of items 3 to 5 and 9 to 12 and in the manner specified in column 4, and
(ii) the information set out in column 1 of items 3 to 34 of the table to section B.29.003, expressed using a description set out in column 2, in micrograms for the information set out in column 1 of items 11, 13, 15, 21, 22, 26, 29, 32 and 33, in micrograms of dietary folate equivalents for the information set out in column 1 of item 20, in milligrams for the information set out in column 1 of items 12, 14, 16 to 19, 23 to 25, 27, 28, 30, 31 and 34, and in grams for the information set out in column 1 of items 3 to 10 and in the manner specified in column 4.
Presentation of Supplemented Food Facts Table
B.29.006 (1) Subject to subsections (2) to (7), the supplemented food facts table must be presented in accordance with the format specified in the applicable figure in the Directory of SFFT Formats, having regard to matters such as order of presentation, dimensions, spacing and the use of upper and lower case letters and bold type.
(2) The characters and rules in the supplemented food facts table must be displayed in a single colour that is a visual equivalent of 100% solid black type on a white background or on a uniform neutral background with a maximum 5% tint of colour.
(3) The characters in the supplemented food facts table
(a) must be displayed in a single standard sans serif font that is not decorative and in such a manner that the characters never touch each other or the rules; and
(b) may be displayed with larger dimensions than those specified in the applicable figure in the Directory of SFFT Formats if all the characters in the table are enlarged in a uniform manner.
(4) The type size shown in parentheses for a version referred to in a table to sections B.29.009 to B.29.015 is the minimum type size that may be used in a supplemented food facts table to show nutrients and supplemental ingredients set out in the tables to sections B.29.002 and B.29.003 in accordance with that version.
(5) A rule that is specified in the applicable figure in the Directory of SFFT Formats as being a 1 point rule or a 2 point rule may be displayed with larger dimensions in the supplemented food facts table.
(6) The information in the supplemented food facts table must be in accordance with subsections B.29.001(2) and (3) and sections B.29.002, B.29.003 and B.29.005.
(7) In a supplemented food facts table consisting of a table in both English and French, the order of languages may be reversed from the order shown in the applicable figure in the Directory of SFFT Formats.
Location of Supplemented Food Facts Table
B.29.007 (1) Subject to subsection (2), the supplemented food facts table must be displayed
(a) in a table in English and a table in French on the same continuous surface of the available display surface;
(b) in a table in both English and French on a continuous surface of the available display surface; or
(c) in a table in English on a continuous surface of the available display surface and a table in French on another continuous surface of the available display surface that is of the same size and prominence as the first surface.
(2) If, in accordance with subsection B.01.012(3), the information required by these Regulations may be shown on the label of a supplemented food in English only or in French only and is shown in that language, the supplemented food facts table may be displayed on the label in a table in that language only on a continuous surface of the available display surface.
Orientation of Supplemented Food Facts Table
B.29.008 (1) Subject to subsection (2), the supplemented food facts table must be oriented in the same manner as other information appearing on the label of a supplemented food.
(2) If a version of a supplemented food facts table cannot be oriented in the same manner as other information appearing on the label, it must be oriented in another manner if there is sufficient space to do so and the food contained in the package does not leak out and is not damaged when the package is turned over.
(3) Subsection (1) does not apply in respect of a supplemented food facts table that is set out on the top or bottom of a package.
Standard and Horizontal Formats
B.29.009 (1) This section applies to a supplemented food unless any of sections B.29.010 to B.29.015 applies to it.
(2) Subject to subsection (3), the supplemented food facts table must be set out in a version listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, a supplemented food facts table in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual standard format in accordance with Figure 3.5(B), 3.6(B) or 3.7(B) of the Directory of SFFT Formats;
(b) the bilingual horizontal format in accordance with Figure 4.3(B), 4.4(B) or 4.5(B) of the Directory of SFFT Formats;
(c) the linear format in accordance with Figures 16.1(E) and (F) or 16.2(E) and (F) of the Directory of SFFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table; or
(e) a manner described in section B.29.017.
(4) For the purposes of this section, in determining whether a version of a supplemented food facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, the supplemented food facts table must include only the information that is required by these Regulations to be included in that table.
(5) Despite subsections (2) and (3), if the supplemented food facts table is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Standard Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 1.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 1.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 1.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 1.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 1.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 1.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Narrow Standard Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 2.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 2.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 2.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 2.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 3
Bilingual Standard Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 3.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 3.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 3.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 3.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 4
Bilingual Horizontal Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 4.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in Parts 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 2 4.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in Parts 1 to 3 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Simplified Formats
B.29.010 (1) This section applies to a supplemented food if it satisfies the condition set out in subsection B.29.002(4) and its supplemented food facts table includes only the information referred to in paragraphs B.29.002(4)(a) to (m).
(2) Subject to subsection (3), the supplemented food facts table must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, a supplemented food facts table containing only the information referred to in paragraphs B.29.002(4)(a) to (m) in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual simplified standard format in accordance with Figure 6.5(B) or 6.6(B) of the Directory of SFFT Formats;
(b) the bilingual simplified horizontal format in accordance with Figure 7.3(B) or 7.4(B) of the Directory of SFFT Formats;
(c) the simplified linear format in accordance with Figures 17.1(E) and (F) or 17.2(E) and (F) of the Directory of SFFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table; or
(e) a manner described in section B.29.017.
(4) Despite subsections (2) and (3), if the supplemented food facts table is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Simplified Standard Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 5.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 5.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 5.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 5.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 5.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 5.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Simplified Standard Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 6.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 6.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 6.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 6.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 3
Bilingual Simplified Horizontal Format
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 7.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in Parts 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 2 7.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in Parts 1 and 2 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Simplified Formats — Supplemented Foods that are Single-serving Prepackaged Products
B.29.011 (1) This section applies to a supplemented food that is a single-serving prepackaged product, whose supplemented food facts table includes only the information referred to in paragraphs B.29.002(5)(a) to (l).
(2) Subject to subsection (3), the supplemented food facts table of the supplemented food that is a single-serving prepackaged product must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(3) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food that is a single-serving prepackaged product, a supplemented food facts table containing only the information referred to in paragraphs B.29.002(5)(a) to (l) in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual simplified standard format in accordance with Figure 6.5.1(B) or 6.6.1(B) of the Directory of SFFT Formats;
(b) the bilingual simplified horizontal format in accordance with Figure 7.3.1(B) or 7.4.1(B) of the Directory of SFFT Formats;
(c) the simplified linear format in accordance with Figures 17.1.1(E) and (F) or 17.2.1(E) and (F) of the Directory of SFFT Formats;
(d) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table; or
(e) a manner described in section B.29.017.
(4) Despite subsections (2) and (3), if the supplemented food facts table of the supplemented food that is a single-serving prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (3)(a), (b) or (c) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Bilingual Simplified Standard Format — Supplemented Foods that are Single-serving Prepackaged Products
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 6.1.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 6.2.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 6.3.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 6.4.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Simplified Horizontal Format — Supplemented Foods that are Single-serving Prepackaged Products
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 7.1.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in Part 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 2 7.2.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in Part 1 and in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Dual Format — Supplemented Foods Requiring Preparation
B.29.012 (1) Subject to subsection (2), if the supplemented food facts table includes information referred to in subsection B.29.005(4), the supplemented food facts table must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, a supplemented food facts table in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual dual format in accordance with Figure 9.5(B) or 9.6(B) of the Directory of SFFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table.
(3) For the purposes of this section, in determining whether a version of a supplemented food facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, the supplemented food facts table must include only the information that is required by these Regulations to be included in the table, together with the information referred to in subsection B.29.005(4).
(4) Despite subsections (1) and (2), if the supplemented food facts table is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Dual Format — Supplemented Foods Requiring Preparation
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 8.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 8.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 8.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 8.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 8.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 8.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Dual Format — Supplemented Foods Requiring Preparation
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 9.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 9.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 9.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 9.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Aggregate Format — Different Kinds of Supplemented Foods
B.29.013 (1) Subject to subsection (2), if the supplemented food facts table of a prepackaged product containing an assortment of supplemented foods includes separate information for each supplemented food as provided in paragraph B.29.005(2)(a) or subsection B.29.005(3), the supplemented food facts table must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, a supplemented food facts table in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out
(a) in the case of a prepackaged product described in paragraph B.29.005(2)(a), in
(i) the bilingual aggregate format in accordance with Figure 11.5(B) or 11.6(B) of the Directory of SFFT Formats,
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table, or
(iii) a manner described in section B.29.017; or
(b) in the case of a prepackaged product described in subsection B.29.005(3), in
(i) the bilingual aggregate format in accordance with Figure 11.5(B) or 11.6(B) of the Directory of SFFT Formats, or
(ii) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table.
(3) For the purposes of this section, in determining whether a version of a supplemented food facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the prepackaged product, the supplemented food facts table must include only the information that is required by these Regulations to be included for each supplemented food for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the supplemented food facts table of the prepackaged product is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in subparagraph (2)(b)(i) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Aggregate Format — Different Kinds of Supplemented Foods
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 10.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 10.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 10.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 10.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 10.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 10.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Aggregate Format — Different Kinds of Supplemented Foods
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 11.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 11.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 11.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 11.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Dual Format — Different Amounts of Supplemented Food
B.29.014 (1) Subject to subsection (2), if the supplemented food facts table includes separate information for different amounts of the supplemented food as provided in paragraph B.29.005(5)(a) without including the information referred to in paragraph B.29.005(5)(b), the supplemented food facts table must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, a supplemented food facts table in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual dual format in accordance with Figure 13.5(B) or 13.6(B) of the Directory of SFFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table.
(3) For the purposes of this section, in determining whether a version of a supplemented food facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, the supplemented food facts table must include only the information that is required by these Regulations to be included for each amount of the supplemented food for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the supplemented food facts table is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Dual Format — Different Amounts of Supplemented Food
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 12.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 12.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 12.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 12.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 12.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 12.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Dual Format — Different Amounts of Supplemented Food
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 13.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 13.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 13.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 13.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Aggregate Format — Different Amounts of Supplemented Food
B.29.015 (1) Subject to subsection (2), if the supplemented food facts table includes separate information for different amounts of the supplemented food as provided in paragraphs B.29.005(5)(a) and (b), the supplemented food facts table must be set out in a version that is listed in column 1 of the table to this section and in respect of which the condition specified in column 2 is satisfied.
(2) If it is not possible to display, in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, a supplemented food facts table in any of the versions that is listed in column 1 of the table to this section, the supplemented food facts table must be set out in
(a) the bilingual aggregate format in accordance with Figure 15.5(B) or 15.6(B) of the Directory of SFFT Formats; or
(b) a version that is listed in column 1 of the table to this section, even though more than 15% of the available display surface would be required to display the supplemented food facts table.
(3) For the purposes of this section, in determining whether a version of a supplemented food facts table cannot be displayed in accordance with these Regulations on 15% or less of the available display surface of the supplemented food, the supplemented food facts table must include only the information that is required by these Regulations to be included for each amount of the supplemented food for which separate information is set out in the table.
(4) Despite subsections (1) and (2), if the supplemented food facts table of the supplemented food is set out on a tag attached to an ornamental container or a tag attached to a package to which a label cannot be physically applied or on which information cannot be legibly set out and easily viewed by the purchaser or consumer under the customary conditions of purchase, it must be set out in a version that is described in paragraph (2)(a) or that is listed in column 1 of the table to this section, without regard to any condition specified in column 2.
TABLE
PART 1
Aggregate Format — Different Amounts of Supplemented Food
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 14.1(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 14.2(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 14.3(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 14.4(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 5 14.5(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 4 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 6 14.6(E) and (F) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 5 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. PART 2
Bilingual Aggregate Format — Different Amounts of Supplemented Food
Item Column 1 Column 2 Figure in Directory of SFFT Formats (Version) Condition of use 1 15.1(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 8 points)
2 15.2(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points)
The version in item 1 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 3 15.3(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 7 points (condensed))
The versions in items 1 and 2 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface. 4 15.4(B) (nutrients and supplemental ingredients to be shown in a type size of not less than 6 points (condensed))
The versions in items 1 to 3 cannot be displayed in accordance with these Regulations on 15% or less of the available display surface.
Presentation of Additional Information
B.29.016 (1) If information referred to in column 1 of the table to section B.29.003 is included in a supplemented food facts table that is set out in a version consisting of a table in English and a table in French or a table in English or French, that information must be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figures 18.1(E) and (F) of the Directory of SFFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of SFFT Formats.
(2) If information referred to in column 1 of the table to section B.29.003 is included in a supplemented food facts table that is set out in a version consisting of a table in both English and French, that information must be displayed
(a) in accordance with the order of presentation, the use of indents and the presentation of footnotes illustrated in Figure 19.1(B) of the Directory of SFFT Formats; and
(b) in respect of matters other than those referred to in paragraph (a), in accordance with the format that is specified in the applicable figure in the Directory of SFFT Formats.
(3) Despite paragraph (1)(a), the use of indents illustrated in Figures 18.1(E) and (F) of the Directory of SFFT Formats is not applicable if information referred to in column 1 of the table to section B.29.003 is set out in the linear format referred to in paragraph B.29.009(3)(c) or the simplified linear format referred to in paragraph B.29.010(3)(c).
Alternative Methods of Presentation
B.29.017 (1) Despite section A.01.016 and subject to subsection (2), a supplemented food facts table that meets the conditions specified in subsection B.29.009(3), B.29.010(3) or B.29.011(3) or paragraph B.29.013(2)(a) may be set out on
(a) a tag attached to the package;
(b) a package insert;
(c) the inner side of a label;
(d) a fold-out label; or
(e) an outer sleeve, overwrap or collar.
(2) The supplemented food facts table must not be set out in a manner described in paragraph (1)(b) or (c) if the label of the supplemented food is required to show a list of cautionary statements.
(3) If the supplemented food facts table is set out in a manner described in paragraph (1)(b) or (c), the outer side of the label of the package must indicate in a type size of not less than 8 points where the supplemented food facts table is located.
(4) If the supplemented food facts table is set out in a manner described in subsection (1), it must be set out
(a) in the case of a supplemented food described in subsection B.29.009(3), in a version that is described in paragraph B.29.009(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.29.009;
(b) in the case of a supplemented food described in subsection B.29.010(3), in a version that is described in paragraph B.29.010(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.29.010;
(c) in the case of a supplemented food described in subsection B.29.011(3), in a version that is described in paragraph B.29.011(3)(a), (b) or (c) or that is listed in column 1 of the table to section B.29.011; and
(d) in the case of a prepackaged product containing an assortment of supplemented foods described in paragraph B.29.013(2)(a), in a version that is described in subparagraph B.29.013(2)(a)(i) or that is listed in column 1 of the table to section B.29.013.
Small Packages
B.29.018 (1) Despite section A.01.016 and subject to subsection (2), if the available display surface of a supplemented food is less than 100 cm2, the label of the supplemented food need not carry a supplemented food facts table if the outer side of the label contains an indication of how a purchaser or consumer may obtain the information that would otherwise be required to be set out in a supplemented food facts table on the label of the supplemented food.
(2) Subsection (1) does not apply to a supplemented food
(a) if the label of the supplemented food is required to show a list of cautionary statements; or
(b) that is the subject of any of the following representations on the label of the supplemented food or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food:
(i) any declaration of the supplemented food’s energy value or the amount of a nutrient or supplemental ingredient contained in the supplemented food,
(ii) any representation that expressly or implicitly indicates that the supplemented food or any substance it contains has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004, and
(iii) any health-related statement, name, logo, symbol, seal of approval or mark.
(3) Despite paragraph (2)(b), subsection (1) applies to a supplemented food that meets the conditions set out in column 2 of item 37 of the Table of Permitted Nutrient Content Statements and Claims for the subject “Free of sugars” set out in column 1 if
(a) the supplemented food is not the subject of any representation referred to in paragraph (2)(b) other than
(i) a statement or claim set out in column 4 of item 37 of the Table of Permitted Nutrient Content Statements and Claims for the subject “Free of sugars” set out in column 1,
(ii) the energy value of the supplemented food expressed in Calories per serving of stated size, and
(iii) the amount of sugar alcohols expressed in grams per serving of stated size;
(b) the statement or claim set out in column 4 of item 37 of the Table of Permitted Nutrient Content Statements and Claims for the subject “Free of sugars” set out in column 1 and that appears on the label is
(i) legibly set out on the principal display panel,
(ii) in lower case letters except for the first letter of each word of the statement or claim, which may be an upper case letter,
(iii) of at least the same size and prominence as the letters used in the numerical portion of the declaration of net quantity as required under paragraph 229(1)(a) and subsections 229(2) and (3) of the Safe Food for Canadians Regulations, and
(iv) displayed in a colour contrasting with the background of the label;
(c) the energy value of the supplemented food expressed in Calories per serving of stated size and the amount of sugar alcohols expressed in grams per serving of stated size are shown immediately after whichever of the following elements appears last on the label:
(i) the list of ingredients,
(ii) a food allergen source, gluten source and added sulphites statement as defined in subsection B.01.010.1(1),
(iii) a declaration referred to in subsection B.01.010.4(1), or
(iv) any statement referred to in subsection B.01.014(1);
(d) the label of the supplemented food is not required to show a list of cautionary statements.
(4) An indication referred to in subsection (1)
(a) must be set out in a type size of not less than 8 points;
(b) must include a postal address, website address or toll-free telephone number; and
(c) must be
(i) in English and French, or
(ii) in one of those languages, if, in accordance with subsection B.01.012(3), the information that is required by these Regulations to be shown on the label of the supplemented food may be shown in that language only and is shown on the label in that language.
(5) The manufacturer of the supplemented food must provide the information referred to in subsection (1) to a purchaser or consumer on request
(a) without charge;
(b) in the following manner, namely,
(i) in the official language in which the information is requested or, if specified by the purchaser or consumer, in both official languages, or
(ii) in one of the official languages, if, in accordance with subsection B.01.012(3), the information that is required by these Regulations to be shown on the label of the supplemented food may be shown in that language only and is shown on the label in that language; and
(c) in the form of a supplemented food facts table that is set out
(i) in a format, other than a horizontal format, that is specified in any of sections B.29.009 to B.29.015 and that would otherwise be carried on the label of the supplemented food in accordance with these Regulations, and
(ii) in a version of that format that is listed in column 1 of item 1 of any Part of the table to the applicable section referred to in subparagraph (i).
B.29.019 If a supplemented food has an available display surface of less than 100 cm2 and has a supplemented food facts table on its label, the supplemented food facts table need only include
(a) the serving of stated size;
(b) the information referred to in column 1 of the table to section B.29.002 or the table to section B.29.003 in respect of the energy value of the supplemented food and the amount of any nutrient or supplemental ingredient that is the subject of a representation on the label of the supplemented food, or in any advertisement for the supplemented food that is made or placed by or on the direction of the manufacturer of the supplemented food, if the representation expressly or implicitly indicates that the supplemented food has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004;
(c) the amount of any added sugar alcohol;
(d) the amount of any saturated fat, sugars or sodium if the amount meets or exceeds the applicable threshold set out in columns 2, 3, 5 or 6 of the table to section B.01.350; and
(e) the amount of any supplemental ingredient for which a cautionary statement set out in the list of cautionary statements is applicable.
Cautionary Statements
B.29.020 (1) Every applicable cautionary statement set out in column 4 of the List of Permitted Supplemental Ingredients in respect of the supplemental ingredients contained in a supplemented food must be shown
(a) in a list on the label of the supplemented food;
(b) grouped together and introduced by the title, in bold type,
(i) “Caution”, “Caution:” or “Caution :” in the English version, and
(ii) “Attention”, “Attention :” or “Attention:” in the French version;
(c) without any intervening printed, written or graphic material appearing between the title and the first cautionary statement;
(d) in regular or bold type;
(e) in lower case letters, except that upper case letters must be used to show the first letter of each cautionary statement; and
(f) separated by a bullet point or comma.
(2) The list of cautionary statements must be shown in a manner that clearly differentiates it on the label by means of one or both of
(a) graphics in the form of a solid-line border around the list or one or more solid lines appearing immediately above, below or at the sides of the list that are the same colour as that of the type used to show the information referred to in subsection B.01.008.1(1); and
(b) a background colour that creates a contrast between the background colour of the list and the background colour used on the adjacent surface of the label, other than the surface used to display
(i) a list of ingredients,
(ii) a food allergen source, gluten source and added sulphites statement as defined in subsection B.01.010.1(1),
(iii) a declaration referred to in subsection B.01.010.4(1),
(iv) any statement referred to in subsection B.01.014(1),
(v) a nutrition facts table, or
(vi) a supplemented food facts table.
(3) The list of cautionary statements must be shown on the label without any intervening printed, written or graphic material,
(a) in English and French, adjacent to each linguistic version of the supplemented food facts table appearing in the same language, if there are separate English and French versions of the table;
(b) in English and French, adjacent to a bilingual version of the supplemented food facts table, with one linguistic version of the list following the other linguistic version; or
(c) in English or French, adjacent to the supplemented food facts table in the same language, if, in accordance with subsection B.01.012(3), the information required by these Regulations to be shown on the label may be shown in that language only and is shown on the label in that language.
(4) If the English and French versions of a list of cautionary statements appear on the label, they must be displayed on a continuous surface of the label’s available display surface, but need not be on the same continuous surface of the label’s available display surface.
(5) If the English and French versions of a list of cautionary statements appear on the same continuous surface of the label, the version that follows the other version must not begin on the same line as that on which the other version ends, unless the available display surface is less than 100 cm2.
Supplemented Food Caution Identifier
Presentation
B.29.021 (1) The principal display panel of a supplemented food must carry the supplemented food caution identifier that is set out in Schedule K.2 if a list of cautionary statements is shown on the label of the supplemented food.
(2) The supplemented food caution identifier must be displayed in black and white and must be in accordance with the identifier set out in Schedule K.2.
(3) Subject to subsection (4), the supplemented food caution identifier must be presented in one of the following formats:
(a) the unilingual standard format, where two separate versions of the identifier are shown, one in English (ES) and one in French (FS); or
(b) the bilingual standard format (BS), where the identifier is shown in both official languages.
(4) If the principal display surface is less than or equal to 450 cm2 and the width of each of the versions of the supplemented food caution identifier in the unilingual standard format or the width of the identifier in the bilingual standard format exceeds the width of the principal display panel, the identifier must be presented in the bilingual compact format (BC) in which the identifier is shown in both official languages.
(5) The supplemented food caution identifier must be displayed in accordance with the applicable specifications set out in the Directory of Supplemented Food Caution Identifier Specifications.
(6) Despite subsection (5), the supplemented food caution identifier may be displayed with larger dimensions than those set out in column 3 of the applicable table in the Directory of Supplemented Food Caution Identifier Specifications if the identifier is enlarged in a proportional manner vertically and horizontally.
(7) If, in accordance with subsection B.01.012(3), the information required by these Regulations may be shown on the label of a supplemented food in English only or in French only and is shown in that language, the supplemented food caution identifier may be displayed on the principal display panel of the supplemented food in that language only on a continuous surface of the available display surface.
(8) If the supplemented food caution identifier is presented in a bilingual format, the order in which the languages appear may be reversed from the order shown in the applicable identifier set out in Schedule K.2.
(9) The characters and other elements of the supplemented food caution identifier must not touch each other.
Placement
B.29.022 (1) The supplemented food caution identifier must be displayed
(a) in the case of a supplemented food with a principal display panel whose height is less than its width, on the right half of the principal display panel; and
(b) in the case of any other supplemented food, on the upper half of the principal display panel.
(2) The supplemented food caution identifier must be surrounded by a buffer that
(a) has a width that is equal to or greater than that set out in column 4 of the applicable table in the Directory of Supplemented Food Caution Identifier Specifications, and
(b) does not contain any text or other graphic material.
(3) If a supplemented food is cylindrical in shape, the outer edge of the buffer must be a minimum distance of 10% of the width of the principal display surface from the edge of the left or right side of that surface.
(4) If it is impossible to comply with both paragraph (1)(a) and subsection (3), the supplemented food caution identifier may be displayed partially in the left half of the principal display panel but only to the extent necessary to comply with that subsection.
Orientation
B.29.023 The supplemented food caution identifier must be oriented in the same manner as most of the other information that appears on the principal display panel unless the panel is displayed in the vertical plane and most of the other information is not displayed parallel with the base of the package, in which case the identifier must be oriented in such a manner that the words appearing in it are parallel with the base.
Prohibitions
B.29.024 It is prohibited to label a prepackaged product with a supplemented food caution identifier or sell a product that is so labelled, unless a list of cautionary statements is shown on the label.
B.29.025 (1) It is prohibited to label a prepackaged product with any representation, including a word, phrase, illustration, sign, mark, symbol or design, that is likely to be mistaken for a supplemented food caution identifier, or sell a prepackaged product that is so labelled.
(2) For the purposes of subsection (1), a representation does not include a nutrition symbol.
Representations
B.29.026 (1) Despite anything in these Regulations, it is prohibited, on the label of or in any advertisement for a supplemented food, to make a statement or claim to the effect that a supplemental ingredient that is a nutrient contained in the supplemented food is generally recognized as an aid in maintaining the functions of the body necessary to the maintenance of good health, if the supplemental ingredient is one for which a cautionary statement set out in the list of cautionary statements is applicable.
(2) However, the label of or advertisement for a supplemented food may carry such a statement or claim if it is accompanied by a statement or claim about the specific action or effect of the supplemental ingredient in maintaining the functions of the body necessary to the maintenance of good health.
(3) If the label of or any advertisement for the supplemented food carries a statement or claim referred to in subsection (1), that statement or claim and the statement or claim referred to in subsection (2) must,
(a) if made on the label of or in an advertisement, other than a radio or television advertisement, for the supplemented food,
(i) be adjacent to one another without any intervening printed, written or graphic material, and
(ii) be shown in letters of the same size and prominence;
(b) if made in a radio advertisement or in the audio portion of a television advertisement for the supplemented food, immediately follow one another; or
(c) if made in the visual portion of a television advertisement for the supplemented food,
(i) appear concurrently and for the same amount of time,
(ii) be adjacent to one another without any intervening printed, written or graphic material, and
(iii) be shown in letters of the same size and prominence.
B.29.027 Despite anything in these Regulations, if a supplemented food is required to show a cautionary statement that is set out in the list of cautionary statements and recommends against consumption by any individual less than 18 years of age that is part of a group identified in the cautionary statement, it is prohibited, on the label of or in any advertisement for the supplemented food, to make a statement or claim to the effect that a nutrient contained in the supplemented food is generally recognized as an aid in maintaining the functions of the body necessary to normal growth and development.
B.29.028 (1) Despite anything in these Regulations, if the label of a supplemented food is required to carry the statement “high caffeine content” in accordance with column 5 of the List of Permitted Supplemental Ingredients, it is prohibited to make a representation, express or implied, on the label of or in any advertisement for the supplemented food with respect to any vitamin or mineral nutrient contained in the supplemented food.
(2) Subsection (1) does not apply in respect of a declaration of a vitamin in a list of ingredients or supplemented food facts table, or in respect of a declaration of a mineral nutrient in a list of ingredients, nutrition symbol or supplemented food facts table.
B.29.029 (1) If a list of cautionary statements is shown on the label of a supplemented food, the representations set out in subsection (2) must meet the following requirements:
(a) if the representation is displayed on the principal display panel,
(i) the height of the upper case letters in the representation must not exceed two times the height of the upper case letters, excluding any accents, in the supplemented food caution identifier, other than in the words “Health Canada” and “Santé Canada”, and
(ii) the height of the tallest ascender of the lower case letters in the representation must not exceed two times the height of the tallest ascender of the lower case letters in the supplemented food caution identifier, other than in the words “Health Canada” and “Santé Canada”; and
(b) if the representation is displayed on any continuous surface, other than on the principal display panel,
(i) the height of the upper case letters in the representation must not exceed two times the height of the upper case letters, excluding any accents, in the cautionary statements, and
(ii) the height of the tallest ascender of the lower case letters in the representation must not exceed two times the height of the tallest ascender of the lower case letters in the cautionary statements.
(2) For the purposes of subsection (1), the representations are the following:
(a) a declaration referred to in subsection B.01.301(1) or (2);
(b) any representation that expressly or implicitly indicates that the supplemented food or any substance it contains has particular nutritional or health-related properties, including any statement or claim set out in column 4 of the Table of Permitted Nutrient Content Statements and Claims or column 1 of the table following section B.01.603 or referred to in section B.01.311, D.01.006 or D.02.004; and
(c) any health-related statement, logo, symbol, seal of approval or mark.
(3) This section does not apply to the brand name or product name of a supplemented food.
Adulteration and Exemptions
B.29.030 A prepackaged product, other than a supplemented food, is adulterated if it contains a supplemented food as an ingredient.
B.29.031 A prepackaged product, other than a supplemented food, is adulterated if a substance listed in column 1 of the List of Permitted Supplemental Ingredients has been added to it other than in accordance with these Regulations.
B.29.032 A supplemented food does not have a poisonous or harmful substance in or on it for the purposes of paragraph 4(1)(a) of the Act — or is not adulterated for the purposes of paragraph 4(1)(d) of the Act — by reason only that a supplemental ingredient has been added to it.
PART CDrugs
DIVISION 1
General
C.01.001 (1) In this Part
- acetaminophen product
acetaminophen product has the same meaning as in Division 9; (produit d’acétaminophène)
- adult standard dosage unit
adult standard dosage unit has, with reference to a drug, the same meaning as in Division 9; (dose normale pour adultes)
- adverse drug reaction
adverse drug reaction means a noxious and unintended response to a drug, which occurs at doses normally used or tested for the diagnosis, treatment or prevention of a disease or the modification of an organic function; (réaction indésirable à une drogue)
- antibiotic
antibiotic means any drug or combination of drugs such as those named in C.01.410 to C.01.592 which is prepared from certain micro-organisms, or which formerly was prepared from micro-organisms but is now made synthetically and which possesses inhibitory action on the growth of other micro-organisms; (antibiotique)
- authorization holder
authorization holder[Repealed, SOR/2017-259, s. 1]
- brand name
brand name means, with reference to a drug, the name, whether or not including the name of any manufacturer, corporation, partnership or individual, in English or French,
(a) that is assigned to the drug by its manufacturer,
(b) under which the drug is sold or advertised, and
(c) that is used to distinguish the drug; (marque nominative)
- case report
case report means a detailed record of all relevant data associated with the use of a drug in a subject; (fiche d’observation)
- children’s standard dosage unit
children’s standard dosage unit has, with reference to a drug, the same meaning as in Division 9; (dose normale pour enfants)
- child resistant package
child resistant package means a package that meets the requirements of subsection (2); (emballage protège-enfants)
- common name
common name means, with reference to a drug, the name in English or French by which the drug is
(a) commonly known, and
(b) designated in scientific or technical journals, other than the publications referred to in Schedule B to the Act; (nom usuel)
- COVID-19
COVID-19 means the coronavirus disease 2019; (COVID-19)
- COVID-19 drug
COVID-19 drug means a drug, other than a veterinary health product, that is manufactured, sold or represented for use in relation to COVID-19; (drogue contre la COVID-19)
- discontinue
discontinue means, in respect of the sale of a drug by the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for the drug, to permanently cease the sale of the drug; (cesser)
- expiration date
expiration date means
(a) in the case of a drug in dosage form, the earlier of the following dates, expressed at minimum as a year and month:
(i) the date up to and including which the drug maintains its labelled potency, purity and physical characteristics, and
(ii) the date after which the manufacturer recommends that the drug not be used; and
(b) in the case of an active ingredient, whichever of the following dates is applicable, expressed at minimum as a year and month:
(i) the retest date, or
(ii) the date after which the manufacturer recommends that the active ingredient not be used; (date limite d’utilisation)
- flavour
flavour means a non-medicinal ingredient or combination of non-medicinal ingredients added to a drug solely to produce or mask a particular taste. It does not include an ingredient or combination of ingredients that impart only a sweet taste to the drug; (saveur)
- fragrance
fragrance means a non-medicinal ingredient or combination of non-medicinal ingredients added to a drug to produce or mask a particular odour; (parfum)
- immediate container
immediate container means the receptacle that is in direct contact with a drug; (récipient immédiat)
- internal use
internal use means ingestion by mouth or application for systemic effect to any part of the body in which the drug comes into contact with mucous membrane; (usage interne)
- ISAD Interim Order
ISAD Interim Order means the Interim Order Respecting the Importation, Sale and Advertising of Drugs for Use in Relation to COVID-19 made by the Minister on September 16, 2020 and published in Part I of the Canada Gazette on October 3, 2020; (arrêté d’urgence IVPD)
- List A
List A means the document, entitled List of Certain Antimicrobial Active Pharmaceutical Ingredients, that is published by the Government of Canada on its website, as amended from time to time; (Liste A)
- List B
List B means the document, entitled List of Certain Veterinary Drugs Which May Be Imported But Not Sold, that is published by the Government of Canada on its website, as amended from time to time; (Liste B)
- List C
List C means the document, entitled List of Veterinary Health Products, that is published by the Government of Canada on its website, as amended from time to time; (Liste C)
- List D
List D means the document entitled List of Certain Non-prescription Drugs for Distribution as Samples that is published by the Government of Canada on its website, as amended from time to time; (Liste D)
- non-medicinal ingredient
non-medicinal ingredient means a substance — other than the pharmacologically active drug — that is added during the manufacturing process and that is present in the finished drug product; (ingrédient non médicinal)
- official drug
official drug means any drug
(a) for which a standard is provided in these Regulations, or
(b) for which no standard is provided in these Regulations but for which a standard is provided in any of the publications mentioned in Schedule B to the Act; (drogue officielle)
- parenteral use
parenteral use means administration of a drug by means of a hypodermic syringe, needle or other instrument through or into the skin or mucous membrane; (usage parentéral)
- per cent
per cent means per cent by weight unless otherwise stated; (pour cent)
- pharmaceutical ink
pharmaceutical ink means a non-medicinal ingredient or combination of non-medicinal ingredients used to imprint the drug with marks or symbols; (encre pharmaceutique)
- pharmacist
pharmacist means a person who
(a) is registered or otherwise entitled under the laws of a province to practise pharmacy, and
(b) is practising pharmacy in that province; (pharmacien)
- pharmacy technician
pharmacy technician means a person who
(a) is registered or otherwise entitled under the laws of a province to practise as a pharmacy technician; and
(b) is practising as a pharmacy technician in that province; (technicien en pharmacie)
- practitioner
practitioner means a person who
(a) is entitled under the laws of a province to treat patients with a prescription drug, and
(b) is practising their profession in that province; (praticien)
- prescription
prescription means an order given by a practitioner directing that a stated amount of any drug or mixture of drugs specified therein be dispensed for the person named in the order; (ordonnance)
- proper name
proper name means, with reference to a drug, the name in English or French
(a) assigned to the drug in section C.01.002,
(b) that appears in bold-face type for the drug in these Regulations and, where the drug is dispensed in a form other than that described in this Part, the name of the dispensing form,
(c) specified in the Canadian licence in the case of drugs included in Schedule C or Schedule D to the Act, or
(d) assigned in any of the publications mentioned in Schedule B to the Act in the case of drugs not included in paragraph (a), (b) or (c); (nom propre)
- salicylate product
salicylate product has the same meaning as in Division 9; (produit de salicylate)
- serious adverse drug reaction
serious adverse drug reaction means a noxious and unintended response to a drug that occurs at any dose and that requires in-patient hospitalization or prolongation of existing hospitalization, causes congenital malformation, results in persistent or significant disability or incapacity, is life-threatening or results in death; (réaction indésirable grave à une drogue)
- serious unexpected adverse drug reaction
serious unexpected adverse drug reaction means a serious adverse drug reaction that is not identified in nature, severity or frequency in the risk information set out on the label of the drug; (réaction indésirable grave et imprévue à une drogue)
- teaspoon
teaspoon means, for the purpose of calculation of dosage, a volume of 5 cubic centimetres; (cuillerée à thé)
- test group
test group means a group that meets the requirements of subsection (3); (groupe d’essai)
- veterinary health product
veterinary health product means any of the following drugs that is in dosage form and that is not manufactured, sold or represented for use in the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms:
(a) a substance set out in Column I of Part 1 of List C that is consistent with the descriptive information set out in Columns II to V, or any combination of any substances in which all the medicinal ingredients are substances set out in Column I of Part 1 of that list if that combination is, in respect of each of those substances, consistent with the descriptive information set out in Columns II and III and the descriptive information set out in Columns IV and V that is, within each of those columns, common to those substances;
(b) a homeopathic medicine set out in Column I of Part 2 of List C that is consistent with the descriptive information set out in Columns II to V, or any combination of homeopathic medicines set out in Column I of Part 2 of that list if that combination is, in respect of each of those homeopathic medicines, consistent with the descriptive information set out in Columns II and III and the descriptive information set out in Columns IV and V that is, within each of those columns, common to those homeopathic medicines; and
(c) a traditional medicine set out in Column I of Part 3 of List C that is consistent with the descriptive information set out in Columns II to V, or any combination of traditional medicines set out in Column I of Part 3 of that list if that combination is, in respect of each of those traditional medicines, consistent with the descriptive information set out in Columns II and III and the descriptive information set out in Columns IV and V that is, within each of those columns, common to those traditional medicines; (produit de santé animale)
- withdrawal period
withdrawal period means the length of time between the last administration of a drug to an animal and the time when tissues or products collected from the treated animal for consumption as food contain a level of residue of the drug that would not likely cause risk to human health. (délai d’attente)
(1.1) For the purposes of the Act, serious adverse drug reaction has the same meaning as in subsection (1).
(2) A child resistant package is a package that
(a) when tested in accordance with an acceptable method,
(i) in the case of a test group comprising children, cannot be opened
(A) by at least 85 per cent of those children prior to a demonstration to them of the proper means of opening the package, and
(B) by at least 80 per cent of those children after the demonstration, and
(ii) in the case of a test group comprising adults
(A) can be opened by at least 90 per cent of those adults, and
(B) where the package is designed so that, once opened and reclosed, it continues to meet the requirements of subparagraph (i), can be so reclosed by at least 90 per cent of those adults; or
(b) complies with the requirements of one of the following standards, namely,
(i) Canadian Standards Association Standard CAN/CSA-Z76.1-M90, entitled Recloseable Child-Resistant Packages, published January 1990, as amended from time to time,
(ii) European Standard EN 28317:1992, entitled Child-resistant packaging — Requirements and testing procedures for reclosable packages, as adopted by the European Committee for Standardization on October 30, 1992, recognized by the British Standards Institution, and effective February 15, 1993 and by the Association française de normalisation, and effective December 20, 1992, and which reiterates fully the international standard ISO 8317:1989, as amended from time to time, and
(iii) Code of Federal Regulations (United States), Title 16, Section 1700.15, entitled Poison prevention packaging standards, as amended from time to time.
(3) For the purposes of this section, test group means
(a) in relation to children, a group of at least 200 children who
(i) are healthy and have no obvious physical or mental disability,
(ii) are between 42 and 51 months of age, and
(iii) represent evenly, within plus or minus 10 per cent, each monthly age between 42 and 51 months calculated to the nearest month; and
(b) in relation to adults, a group of at least 100 adults who
(i) are healthy and have no obvious physical or mental disability,
(ii) are between 18 and 45 years of age, and
(iii) represent evenly, within plus or minus 10 per cent, each yearly age between 18 and 45 years calculated to the nearest year.
(4) For the purpose of this section, an amendment from time to time to a standard referred to in paragraph (2)(b) becomes effective 18 months after the date designated by the competent authority as the effective date for the amendment.
- SOR/80-543, s. 1
- SOR/85-966, s. 1
- SOR/86-93, s. 1
- SOR/87-484, s. 1
- SOR/92-654, s. 1
- SOR/93-202, s. 1
- SOR/95-411, s. 1
- SOR/95-521, s. 1
- SOR/96-399, s. 1
- SOR/96-240, s. 1
- SOR/97-543, s. 5
- SOR/2010-105, s. 1
- SOR/2013-74, s. 1
- SOR/2013-122, s. 3
- SOR/2016-139, s. 1
- SOR/2017-76, s. 1
- SOR/2017-133, s. 1
- SOR/2017-259, s. 1
- SOR/2020-74, s. 1
- SOR/2021-45, s. 1
- SOR/2021-46, s. 11(E)
- SOR/2022-197, s. 8
- SOR/2023-247, s. 1(F)
C.01.001A [Repealed, SOR/98-423, s. 1]
C.01.002 The Proper Name of a drug shown opposite an item number in the following Table in the column headed “Chemical Names and Synonyms” shall be the name shown opposite that item number in the column headed “Proper Names”.
TABLE
| Item No. | Proper Names | Chemical Names and Synonyms |
|---|---|---|
| A.1 | Acepromazine | 2-acetyl-10-(3-dimethylaminopropyl) iephenothiazine |
| A.2 | Acetaminophen | p-Acetaminophenol, Paracetamol, p-Hydroxyacetanilide: N-acetyl-p-aminophenol |
| A.3 | Acetanilide: Acetanilid | Acetylaminobenzene: Antifebrin: Phenylacetamide |
| A.4 | Acetylsalicylic Acid | Acetylsalicylic acid |
| A.5 | Allopurinol | 1-H-Pyrazolo [3,4-d] pyrimidin-4-ol: 4-Hydroxypyrazolo (3,4-d) pyrimidine |
| A.6 | Amantadine | 1-Adamantanamine |
| A.7 | Aminocaproic acid | 6-Aminohexanoic acid |
| A.8 | Aminopterin | N-[4-(2,4-diamino-6-pteridyl methyl) amino-benzoyl]-L- glumatic acid |
| A.9 | Aminopyrine: Amidopyrine | 1,5-dimethyl-2-phenyl-4-dimethylamino-3-pyrazolone: Dimethylaminophenazone |
| A.10 | Amitriptyline | 3-(3-Dimethylaminopropylidene)-1,2: 4,5-dibenzocyclohepta-1,4-diene |
| A.11 | Azacyclonol | α,α-diphenyl-4-piperidinecarbinol |
| B.1 | Bemegride | 3-Ethyl-3-methylglutarimide |
| B.2 | Benactyzine | Dimethylaminoethyl-1,1-diphenylglycolate |
| B.3 | Bendroflumethiazide | 3-benzyl-3,4-dihydro-6-(trifluoro- methyl)-2H-1,2,4-benzothiadiazine-7-sulfonamide-1,1-dioxide: Bendrofluazide (B.A.N.) |
| B.4 | Betahistine | 2-[2-(Methylamino)ethyl] pyridine |
| B.5 | Bethanidine | N-Benzyl-N′N″-dimethylguanidine: 1-Benzyl-2,3-dimethylguanidine |
| B.6 | Bretylium tosylate | N-2-Bromobenzyl-N-ethyl-N, N-dimethylammonium tosylate (Tosylic acid is trivial name for p-toluenesulphonic acid) |
| B.7 | Bromisoval | 2-monobromoisovalerylurea: Bromisovalum: Bromvalitone |
| C.1 | Calcium Carbimide | Calcium cyanamide |
| C.2 | Captodiamine | 4-butylthio-α-phenylbenzyl-2- dimethylaminoethylsulfide |
| C.3 | Carisoprodol | N-Isopropyl-2-methyl-2-propyl-1, 3-propanediol dicarbamate |
| C.4 | Carphenazine | 1-[10-(3[4-(2-Hydroxyethyl)-1-piperazinyl]propyl) phenothiazin-2yl]-1-propapone |
| C.5 | Cephaloridine | 7-[(2-Thienyl) acetamido]-3-(1-pyridylmethyl)-3-cephem-4-carboxylic acid betaine |
| C.6 | Chlormezanone | 2-(4-chlorophenyl)-3-methyl-4-methathiazanone-1,1-dioxide: Chlormethazone: Chlormethazanone |
| C.7 | Chloromethapyrilene | N,N-dimethyl-N′-(2-pyridyl)-N′-(5-chloro-2-thenyl)-ethylenediamine: Chlorothen |
| C.8 | Chlorphentermine | 4-Chloro-α,α-dimethylphenethylamine |
| C.9 | Cinchocaine | 2-butoxy-N-(2-diethylaminoethyl) cinchoninamide: Dibucaine |
| C.10 | Cinchophen | 2-phenylquinoline-4-carboxylic acid: Quinophan |
| C.11 | Clofibrate | Ethyl 2-(p-chlorophenoxy)-2-methylpropionate |
| C.12 | Clomiphene | 1-Chloro-2-[4-(2-diethylamino-ethoxy)phenyl]-1,2-diphenylethylene: 2-[p-(2-Chloro-1,2-diphenylvinyl)phenoxy] triethylamine |
| D.1 | Desipramine | 5-(3-Methylaminopropyl)-10,11-dihydro-5H-dibenz[b,f]azepine |
| D.2 | Diazepam | 7-Chloro-1,3-dihydro-1-methyl-5-phenyl-2H-1,4-benzodiazepin-2-one |
| D.3 | Diethylpropion | 1-phenyl-2-diethylaminopropanone-1 |
| D.4 | Diphenidol | 1,1-Diphenyl-4-piperidinobutan-1-ol |
| D.5 | Disulfiram | Tetraethylthiuram disulphide |
| E.1 | Ectylurea | 2-ethyl-cis-crotonylurea |
| E.2 | Emylcamate | 1-Ethyl-1-methylpropyl carbamate |
| E.3 | Ethacrynic Acid | [2,3-Dichloro-4-(2-methylenebutyryl) phenoxy] acetic acid: 2,3-Dichloro-4-(2-ethylacryloyl) phenoxyacetic acid |
| E.4 | Ethchlorvynol | 3-(2-chlorovinyl)-1-pentyn-3-ol |
| E.5 | Ethinamate | 1-ethynylcyclohexyl carbamate |
| E.6 | Ethionamide | 2-Ethylisonicotinthioamide |
| E.7 | Ethomoxane | 2-n-Butylaminomethyl-8-ethoxy-benzo-1,4-dioxan |
| E.8 | Ethyl Trichloramate | Ethyl n-[1-(2,2,2,-trichloro-1-hydroxyethyl)] carbamate |
| E.9 | Etryptamine | 3-(2-Aminobutyl) indole |
| E.10 | Etymemazine | 10-(3-Dimethylamino-2-methylpro-pyl)-2-ethylphenothiazine |
| F.1 | Fluphenazine | 10-{3-[4-(2-Hydroxyethyl) piperazin-1-yl] propyl}-2-tri-fluoromethylphenothiazine |
| F.2 | Furosemide | 4-Chloro-N-furfuryl-5-sulphamoylanthranilic acid: Frusemide (B.A.N.) |
| G.1 | Glyburide | 5-chloro-N-[2-[4-[[[(cyclohexylamino carbonyl]amino]sulfonyl]phenyl]ethyl]-2-methoxy benzamide: 1-4[4-[2-(5-chloro-2-methoxybenzamido)ethyl]phenyl- sulfonyl]-3-cyclohexylurea: Glibenclamide |
| H.1 | Haloperidol | 4-(4-Chlorophenyl)-1-[3-(4-fluorobenzoyl) propyl]-piperidin-4-ol: 4-[4-(p-Chlorophenyl)-4-hydro-xypiperidino]-4’-fluorobutyro-phenone |
| H.2 | Hydroxychloroquine | 7-Chloro-4[4-(N-ethyl-N-2-hydro-xyethylamino)-1-methylbutyl-amino] quinoline |
| H.3 | Hydroxyzine | 1-(p-chloro-α-phenylbenzyl)-4-(2-hydroxy ethoxyethyl) piperazine |
| I.1 | Idoxuridine | 5-Iodo-2′-deoxyuridine |
| I.2 | Imipramine | 5-(3-dimethylaminopropyl)-10,11-dihydro-5H-dibenz[b,f]azepine |
| I.3 | Indomethacin | 1-(p-Chlorobenzoyl)-5-methoxy-2-methyl-indole-3-acetic acid |
| I.4 | Iproniazid | 1-isonicotinoyl-2-isopropylhydrazine |
| I.5 | Isocarboxazid | 3-N-Benzylhydrazinocarbonyl-5-methylisoxazole |
| I.6 | Isoproterenol | 3,4-Dihydroxy-α-[isopropylamino) methyl] benzyl alcohol: Isoprenaline |
| L.1 | Liothyronine | L-α-Amino-3-[(4-hydroxy-3-iodophenoxy)-3,5-di-iodo-phenyl] propionic acid |
| M.1 | Mefenamic acid | N-(2,3-Xylyl)-anthranilic acid |
| M.2 | Melphalan | 4-Di-(2-chlorethyl)amino-L-phenylalanine |
| M.3 | Mepazine | 10-[(1-methyl-3-piperidyl) methyl] phenothiazine |
| M.4 | Mephenesin | 3-o-toloxy-1,2-propanediol |
| M.5 | Mephenoxalone | 5-(o-Methoxyphenoxymethyl)-2-oxazolidinone |
| M.6 | Meprobamate | 2,2-di(carbamoylmethyl) pentane |
| M.7 | Methaqualone | 2-Methyl-3-o-tolyquinazolin-4-one: 2-Methyl-3-o-tolyl-4-quinazolone |
| M.8 | Methisazone | 1-Methylindoline-2,3-dione-3-thiosemicarbazone: N-Methylisatin-ß-thiosemicarbazone |
| M.9 | Methotrimeprazine | 10-[3-(2-Methyl)dimethylamino propyl]-2-methoxyphenothiazine: Levomepromazine |
| M.10 | Methyldopa | 1-3(3,4-Dihydroxyphenyl)-2-methylalanine |
| M.11 | Methylparafynol | 3-methyl-1-pentyn-3-ol: Methylpentynol |
| M.12 | Methylphenidate | Methyl-1-phenyl-1-(2-piperidyl) acetate |
| M.13 | Methyprylon | 3,3-diethyl-5-methyl-2,4-piperidinedione |
| M.14 | Methysergide | 1-(Hydroxymethyl)propylamide of 1-methyl-d-lysergic acid |
| M.15 | Metyrapone | 2-Methyl-1,2-di(3-pyridyl)propan-1-one |
| N.1 | Nalidixic Acid | 1-Ethyl-7-methyl-4-oxo-1,8-naphthyridine-3-carboxylic acid |
| N.2 | Nialamide | 1-[2-(benzycarbamyl)ethyl]-2-isonicotinoyl-hydrazine |
| N.3 | Nortriptyline | 3-(3-Methylaminopropylidene)-1,2, 4,5-dibenzocyclohepta-1,4-diene |
| O.1 | Oxanimide | 2-ethyl-3-propyl-glycidamide |
| O.2 | Oxazepam | 7-Chloro-1,3-dihydro-3-hydroxy-5-phenyl-1,4-benzodiazepin-2-one |
| O.3 | Oxyphenbutazone | 4-n-Butyl-2-(4-hydroxyphenyl)-1-phenyl-pyrazolidine-3,5-dione |
| P.1 | Paramethadione | 3,5-dimethyl-5-ethyl-2,4-oxazolidinedione |
| P.2 | Pargyline | N-Benzyl-N-methylprop-2-ynylamine |
| P.3 | Pemoline | 2-Imino-5-phenyloxazolidin-4-one |
| P.4 | Pentazocine | 1,2,3,4,5,6-Hexahydro-8-hydroxy-6,11-dimethyl-3-(3-methylbut-2-enyl)-2,6-methano-3-benzazocine: 1,2,3,4,5,6-Hexahydro-6,11-dimethyl-3-(3-methyl-2-butenyl)-2,6-methano-3-benzazocin-8-ol |
| P.5 | Pentolinium Tartrate | NN′-Pentamethylenedi-(methylpryrrolidinium hydrogen, tartrate) |
| P.6 | Perphenazine | 2-chloro-10-{3-[1-(2-hydroxyethyl)-4-piperazinyl]propyl} phenothiazine |
| P.7 | Phacetoperane | l-1-Phenyl-1(2′-piperidyl)-1-acetoxymethane |
| P.8 | Phenacemide | (Phenylacetyl)urea |
| P.9 | Phenacetin | p-acetphenetidin: Acetphenetidin: Acetophenetidin: p-ethoxyacetanilid |
| P.10 | Phenaglycodol | 2-p-chlorophenyl-3-methyl-2,3-butanediol |
| P.11 | Phendimetrazine | 3,4-Dimethyl-2 Phenylmorpholine |
| P.12 | Phenelzine | 2-phenylethylhydrazine |
| P.13 | Phenformin | N′-ß-phenethylformamidinyliminourea |
| P.14 | Pheniprazine | α-Methylphenethylhydrazine |
| P.15 | Phenmetrazine | Tetrahydro-3-methyl-2-phenyl-1,4-oxazine: 3-methyl-2-phenylmorpholine |
| P.16 | Phentermine | α, α-Dimethylphenethylamine: phenyl-tert-butylamine |
| P.17 | Phenylindanedione | 2-phenylindane-1,3-dione |
| P.18 | Phenyltoloxamine | N,N-dimethyl-2-(α-phenyl-o-tolyloxy) ethylamine |
| P.19 | Pholedrine | p-(4-hydroxyphenyl)-isopropylmethylamine |
| P.20 | Piperliate | 1-piperidine-ethanol benzilate |
| P.21 | Pipradol | Diphenyl-2-piperidylmethanol |
| P.22 | Prochlorperazine | 2-Chloro-10-[3-(1-methyl-4-piperazinyl) propyl]phenothiazine |
| P.23 | Prodilidine | 1,2-Dimethyl-3-phenyl-3-pyrrolidinyl propionate |
| P.24 | Propranolol | 1-(Isopropylamino)-3-(1-naphthyloxy)-2-propanol |
| P.25 | Prothipendyl | 9-(3-Dimethylaminopropyl)-10-thia-1,9-diaza-anthracene |
| P.26 | Protriptyline | 7-(3-Methylaminopropyl)-1,2:5,6-dibenzocycloheptatrien: N-Methyl-5H-dibenzo [a, d] cycloheptene-5-propylamine |
| P.27 | Pyrazinamide | Pyrazinoic acid amide |
| R.1 | Rifampin | 3-{[(4-methyl-1-piperazinyl)imino]methyl} rifamycin SV : Rifampicin (I.N.N.) (Rifamycin SV is an antibiotic produced by Streptomyces mediterranei) |
| S.01 | Sodium Cromoglycate | 4H-1-Benzopyran-2-carboxylic acid, 5,5′-[(2-hydroxy-1,3-propanediyl) bis(oxy)]bis[4-oxo-,disodium salt]: |
| Disodium 5,5′-(2-hydroxytrimethylenedioxy) bis[4-oxo-4H-1-benzopyran-2- carboxylate]: Disodium 4,4′-dioxo-5,5′-(2-hydroxytrimethylenedioxy)di (chromene-2-carboxylate): Cromolyn Sodium (USP): Disodium Cromoglycate | ||
| S.1 | Sulfameter | 2-(4-Aminobenzenesulphonamido)-5-methoxypyrimidine: N′-(5-methoxy-2-pyrimidinyl) sulfanilamide: Sulfamethoxydiazine (B.A.N.) |
| S.2 | Sulfamethazine | N′-(4,6-dimethyl-2- pyrimidyl)sulfanilamide: 2-(p-aminobenzenesulphonamide)-4,6-dimethylpyrimidine: sulphadimedine |
| S.3 | Sulfinpyrazone | 1,2-diphenyl-4-(2-phenylsulfinilethyl)-3,5-pyrazolidinedione |
| S.4 | Sulfisoxazole | 3,4-dimethyl-5-sulfanilamidoisoxazole: Sulphafurazole |
| T.1 | Tetracaine | 2-dimethylaminoethyl-p-n- butylaminobenzoate: Amethocaine |
| T.2 | Thiethylperazine | 2-Ethylthio-10-[3-(4- methylpiperazin-1-yl) propyl]phenothiazine |
| T.3 | Thiopropazate | 2-chloro-10-[3-[1-(2-acetoxyethyl)-4-piperazinyl] propyl]phenothiazine |
| T.4 | Thioproperazine | 2-Dimethylsulphamoyl-10-[3-(4-methylpiperazin-1-yl)- propyl]phenothiazine |
| T.5 | Thioridazine | 10-{2-[2-(1-methylpiperidyl)] ethyl α}-2-methylthiopheno- thiazine |
| T.6 | Tranylcypromine | Trans d, 1-2-phenylcyclopropyl- amine |
| T.7 | Triamterene | 2,4,7-Triamino-6-phenylpteridine |
| T.8 | Triflupromazine | 10-(3-dimethylaminopropyl)-2-trifluoromethylphenothiazine: Fluopremazine |
| T.9 | Trimeprazine | 10-(3-dimethylamino-2-methylpropyl) phenothiazine |
| T.10 | Trimethadione | 3,5,5-trimethyl-2,4-oxazolidine- dione: Troxidone |
| T.11 | Trimipramine | 5-(3-Dimethylamino-2-methylpropyl)-10,11-dihydro-5H-dibenz[b,f]azepine: 5-(3′-Dimethylamino-2′-methylpropyl)iminodibenzyl |
| T.12 | Tybamate | 2-Methyl-2-propyltrimethylene butylcarbamate carbamate: 2-(Hydroxymethyl)-2-methyl-pentyl butylcarbamate carbamate |
| V.1 | Vinblastine | An alkaloid derived from Vinca rosea |
| V.2 | Vincristine | An alkaloid derived from Vinca rosea |
- SOR/87-565, s. 1
- SOR/88-182, s. 1
- SOR/88-482, s. 1(F)
- SOR/90-173, s. 1(F)
C.01.003 No person shall sell a drug that is not labelled as required by these Regulations.
- SOR/80-544, s. 1
C.01.004 (1) The inner and outer labels of a drug shall show
(a) on the principal display panel
(i) the proper name, if any, of the drug which, if there is a brand name for the drug, shall immediately precede or follow the brand name in type not less than one-half the size of that of the brand name,
(ii) if there is no proper name, the common name of the drug,
(iii) where a standard for the drug is prescribed in Division 6 of this Part, a statement that the drug is a Canadian Standard Drug, for which the abbreviation C.S.D. may be used,
(iv) where a standard for the drug is not prescribed in Division 6 of this Part but is contained in a publication mentioned in Schedule B to the Act, the name of the publication containing the standard used or its abbreviation as provided in Schedule B or, if a manufacturer’s standard is used, a statement setting forth the fact that such a standard is used, and
(v) in both official languages, the notation “sterile” “stérile” if the drug is required to be sterile by these Regulations;
(b) on the upper left quarter of the principal display panel
(i) the symbol “Pr” in the case of a prescription drug, but the symbol “Pr” shall not appear on the label of any other drug,
(ii) the symbol “
 ” in a clear manner and a conspicuous colour and size, in the case of a controlled drug, other than a controlled drug contained in an agricultural implant and set out in Part III of the schedule to Part G,
” in a clear manner and a conspicuous colour and size, in the case of a controlled drug, other than a controlled drug contained in an agricultural implant and set out in Part III of the schedule to Part G,(iii) the symbol “N” in a colour contrasting with the rest of the label or in type not less than half the size of any letters used thereon, in the case of a narcotic as defined in the Narcotic Control Regulations, and
(iv) in the case of a targeted substance as defined in section 1 of the Benzodiazepines and Other Targeted Substances Regulations, the following symbol in a colour contrasting with the rest of the label and in type not less than half the size of any other letter used on the main panel, namely,

(c) on any panel
(i) the name and address of the manufacturer of the drug,
(ii) the lot number of the drug,
(iii) adequate directions for use of the drug, except in the case of a drug to which section C.01.004.02 applies,
(iv) a quantitative list of the medicinal ingredients of the drug by their proper names or, if they have no proper names, by their common names, except in the case of a drug to which section C.01.004.02 applies,
(v) the expiration date of the drug, and
(vi) in the case of a new drug for extraordinary use in respect of which a notice of compliance has been issued under section C.08.004.01, the following statement, displayed in capital letters and in a legible manner:
“HEALTH CANADA HAS AUTHORIZED THE SALE OF THIS EXTRAORDINARY USE NEW DRUG FOR [naming purpose] BASED ON LIMITED CLINICAL TESTING IN HUMANS.
SANTÉ CANADA A AUTORISÉ LA VENTE DE CETTE DROGUE NOUVELLE POUR USAGE EXCEPTIONNEL AUX FINS DE [indication de la fin] EN SE FONDANT SUR DES ESSAIS CLINIQUES RESTREINTS CHEZ L’ÊTRE HUMAIN.”.
(1.1) to (1.5) [Repealed, SOR/2014-158, s. 3]
(2) In addition to the requirements of subsection (1), the outer label of a drug shall display the following information:
(a) the net amount of the drug in the container in terms of weight, measure or number;
(b) in the case of a drug intended for parenteral use, a quantitative list of any preservatives present therein by their proper names or, if they have no proper names, by their common names; and
(c) in the case of a drug for human use that contains mercury or a salt or derivative thereof as a preservative, a quantitative list of all mercurial preservatives present therein by their proper names or, if they have no proper names, by their common names.
(3) Where the container of a drug is too small to accommodate an inner label that conforms to the requirements of these Regulations, the inner label requirements of these Regulations do not apply to the drug in that container if
(a) there is an outer label that complies with the labelling requirements of these Regulations; and
(b) the inner label shows
(i) the proper name of the drug, the common name of the drug if there is no proper name or, in the case of a drug with more than one medicinal ingredient, the brand name of the drug,
(ii) the potency of the drug except where, in the case of a drug with more than one medicinal ingredient, the name used pursuant to subparagraph (i) for that drug is unique for a particular potency of the drug,
(iii) the net contents of the drug if it is not in a discrete dosage form,
(iv) the route of administration of the drug if other than oral,
(v) the lot number of the drug,
(vi) the name of the manufacturer of the drug,
(vii) the expiration date of the drug, and
(viii) the identification of special characteristics of the dosage form if they are not evident from the name of the drug under subparagraphs (i) or (ii).
(4) [Repealed, SOR/92-654, s. 2]
(5) This section does not apply to
(a) a drug sold to a drug manufacturer; or
(b) a drug dispensed pursuant to a prescription, if its label carries adequate directions for use and complies with the requirements of section C.01.005.
- SOR/80-543, s. 2
- SOR/81-334, s. 1(E)
- SOR/85-715, s. 2
- SOR/89-229, s. 1
- SOR/90-216, s. 1
- SOR/90-586, s. 1
- SOR/92-654, s. 2
- SOR/93-202, s. 2
- SOR/97-228, s. 1
- SOR/97-515, s. 1
- SOR/2000-219, s. 1
- SOR/2001-181, s. 4
- SOR/2010-105, s. 2
- SOR/2011-88, s. 1
- SOR/2013-122, ss. 4, 5
- SOR/2014-158, s. 3
- SOR/2019-170, s. 18
- SOR/2023-247, s. 2
C.01.004.01 (1) Every label of a drug for human use in dosage form shall display the following:
(a) a telephone number, email address, website address, postal address or any other information that enables communication with a contact person in Canada; and
(b) a statement to the effect that any injury to a person’s health that is suspected of being associated with the use of the drug may be reported to the contact person.
(2) Subsection (1) does not apply to
(a) the labels of a drug that is listed in Schedule C or D to the Act and that is in dosage form; or
(b) the inner and outer labels of a drug to which section C.01.004.02 applies.
- SOR/2014-158, s. 4
C.01.004.02 (1) In addition to the requirements of section C.01.004, the outer label of a drug for human use in dosage form shall display, either one bilingual table, placed on any panel, that contains only the following information in both English and French or one table in English and one table in French, each of which is placed on any panel, that contains only the following information:
(a) adequate directions for use of the drug;
(b) a quantitative list of the drug’s medicinal ingredients by their proper names or, if they have no proper names, by their common names;
(c) the drug’s non-medicinal ingredients listed in alphabetical order or in descending order of predominance by their proportion in the drug, preceded by words that clearly distinguish them from the medicinal ingredients; and
(d) the information referred to in subsection C.01.004.01(1).
(2) If a package is too small to accommodate an outer label that displays one bilingual table that lists all of the drug’s non-medicinal ingredients or two unilingual tables, each of which lists all of the drug’s non-medicinal ingredients, the list of non-medicinal ingredients shall be displayed in both English and French on a tag, tape or card that is attached to the package.
(3) If pharmaceutical ink, a fragrance or a flavour has been added to the drug, the following expressions may be included in the list of non-medicinal ingredients to indicate that those ingredients have been added to the drug, instead of listing them individually:
(a) in the case where a bilingual table referred to in subsection (1) is displayed, the expressions “flavour/saveur”, “fragrance/parfum” and “pharmaceutical ink/encre pharmaceutique”; or
(b) in the case where two unilingual tables referred to in subsection (1) are displayed, the expressions
(i) “encre pharmaceutique”, “parfum” and “saveur” in the table in French, and
(ii) “flavour”, “fragrance” and “pharmaceutical ink” in the table in English.
(4) If the composition of the drug varies from one lot to another with respect to its non-medicinal ingredients,
(a) in the case where a bilingual table referred to in subsection (1) is displayed, the table shall include a reference to all non-medicinal ingredient alternatives that may be present in the drug, preceded by the symbol “+/–” or “±” or the expression “or/ou” or “may contain/peut contenir”; or
(b) in the case where two unilingual tables referred to in subsection (1) are displayed,
(i) the table in French shall list all non-medicinal ingredient alternatives that may be present in the drug, preceded by the symbol “+/–” or “±” or the expression “ou” or “peut contenir”, and
(ii) the table in English shall list all non-medicinal ingredient alternatives that may be present in the drug, preceded by the symbol “+/–” or “±” or the expression “or” or “may contain”.
(5) For the purposes of paragraphs (3)(a) and (4)(a), the French terms in the expressions may appear first.
(6) Subsections (1) to (5) do not apply to
(a) prescription drugs;
(b) drugs that are permitted to be sold without a prescription but that are to be administered only under the supervision of a practitioner; and
(c) drugs that are represented as being solely for use as a disinfectant on hard non-porous surfaces.
- SOR/2014-158, s. 5
- SOR/2017-18, s. 23
- SOR/2018-69, s. 13
- SOR/2021-46, s. 5
C.01.004.03 In addition to the requirements of section C.01.004, the inner label of a drug to which section C.01.004.02 applies shall display on any panel
(a) adequate directions for use of the drug;
(b) a quantitative list of the drug’s medicinal ingredients by their proper names or, if they have no proper names, by their common names; and
(c) the information referred to in subsection C.01.004.01(1).
- SOR/2014-158, s. 5
C.01.004.1 (1) No person shall import a drug in dosage form into Canada for the purpose of sale unless they have in Canada a person who is responsible for the sale of the drug.
(2) No person who imports a drug in dosage form into Canada shall sell any lot or batch of the drug unless the name of the person who imports it, and the address of the principal place of business in Canada of the person responsible for its sale, appears on the inner and outer labels of the drug.
- SOR/82-524, s. 1
- SOR/93-475, s. 1
- SOR/97-12, s. 2
C.01.005 (1) The principal display panel of both the inner label and outer label of a drug in dosage form shall show the drug identification number assigned for that drug, preceded by the expression “Drug Identification Number” or “Drogue : identification numérique”, or both, or the abbreviation “DIN”.
(2) Subsection (1) does not apply to
(a) a drug in dosage form that is compounded by a pharmacist under a prescription or by a practitioner; or
(b) a drug in dosage form that is sold under a prescription if the following information appears on the drug’s label:
(i) the drug’s proper name, common name or brand name,
(ii) the drug’s potency, and
(iii) the name of the drug’s manufacturer.
(3) In this section and in sections C.01.005.1 and C.01.014, drug in dosage form means a drug in a form in which it is ready for use by the consumer without requiring any further manufacturing.
(4) and (5) [Repealed, SOR/81-248, s. 1]
- SOR/81-248, s. 1
- SOR/93-202, s. 3
- SOR/98-423, s. 2
- SOR/2001-181, s. 4
- SOR/2017-259, s. 2
- SOR/2018-69, s. 27
- SOR/2018-77, s. 1
C.01.005.1 (1) No pharmacist or practitioner shall sell a Class A opioid — including one that is compounded by a pharmacist under a prescription or by a practitioner — unless
(a) the drug’s package has applied to it a warning sticker that meets the specifications set out in the source document; and
(b) the drug is accompanied by a patient information handout that meets the specifications set out in the source document.
(2) Subsection (1) does not apply in respect of the sale of a Class A opioid by a pharmacist or practitioner if
(a) the opioid will be, or is, administered under the supervision of a practitioner; or
(b) the sale is to a pharmacist or practitioner.
(3) The following definitions apply in this section.
- Class A opioid
Class A opioid means a drug in dosage form set out in Part A of the List of Opioids, published by the Government of Canada on its website, as amended from time to time. (opioïde de catégorie A)
- source document
source document means the document entitled Information for Patients Concerning Opioids, published by the Government of Canada on its website, as amended from time to time. (document source)
- SOR/2018-77, s. 2
C.01.006 Where a package of a drug has only one label, that label shall contain all the information required by these Regulations to be shown on both the inner and the outer labels.
C.01.007 No reference, direct or indirect, to the Act or to these Regulations shall be made upon any label of or in any advertisement for a drug unless such reference is a specific requirement of the Act or these Regulations.
C.01.008 [Repealed, SOR/80-544, s. 2]
C.01.009 If any Act of Parliament or any of its regulations prescribes a standard or grade for a drug and that standard or grade is given a name or designation in the Act or regulation, no person shall, on a label of or in any advertisement for that drug, use that name or designation unless the drug conforms with the standard or grade.
- SOR/2017-18, s. 13
C.01.010 If it is necessary to provide adequate directions for the safe use of a parenteral drug or prescription drug that is used in the treatment or prevention of any disease, disorder or abnormal physical state mentioned in Schedule A.1 to the Act, the disease, disorder or abnormal physical state may be mentioned on the drug’s labels, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the drug, and, in that respect, the drug is exempt from subsections 3(1) and (2) of the Act.
- SOR/2013-122, s. 6
- SOR/2014-158, s. 6
- SOR/2017-18, s. 21(F)
- SOR/2018-69, s. 14(F)
- SOR/2021-46, s. 10
C.01.011 (1) A drug referred to in subsection 10(2) of the Act shall be exempt from the standard for any drug contained in any publication mentioned in Schedule B to the Act to the extent that such drug differs from that standard with respect to colour, flavour, shape and size, if such difference does not interfere with any method of assay prescribed in any such publication.
(2) [Repealed, SOR/93-243, s. 2]
(3) Where a manufacturer’s standard is used for a drug, the manufacturer shall make available to the Minister, on request, details of that standard and of a method of analysis for the drug acceptable to the Minister.
(4) No person shall use a manufacturer’s standard for a drug that provides
(a) a lesser degree of purity than the highest degree of purity, or
(b) a greater variation in potency than the least variation in potency,
provided for that drug in any publication mentioned in Schedule B to the Act.
- SOR/93-243, s. 2
- SOR/2018-69, ss. 31(E), 32(F)
C.01.012 A manufacturer who makes representations on a label of a drug in oral dosage form, or in any advertisement, with respect to the site, rate or extent of release to the body of a medicinal ingredient of the drug, or the availability to the body of a medicinal ingredient of the drug, shall
(a) before making the representations, conduct such investigations, using an acceptable method, as may be necessary to demonstrate that the site, rate or extent of release to the body of the medicinal ingredient of the drug and the availability to the body of the medicinal ingredient of the drug, correspond to the representations; and
(b) on request submit the record of such investigations to the Minister.
- SOR/89-455, s. 2
- SOR/94-36, s. 1
- SOR/2018-69, s. 27
C.01.013 (1) Where the manufacturer of a drug is requested in writing by the Minister to submit on or before a specified day evidence with respect to a drug, the manufacturer shall make no further sales of that drug after that day unless he has submitted the evidence requested.
(2) If the Minister determines that the evidence submitted by a manufacturer under subsection (1) is not sufficient, he or she shall so notify the manufacturer in writing.
(3) Where, pursuant to subsection (2), a manufacturer is notified that the evidence with respect to a drug is not sufficient, he shall make no further sales of that drug unless he submits further evidence and is notified in writing by the Minister that that further evidence is sufficient.
(4) A reference in this section to evidence with respect to a drug means evidence to establish the safety of the drug under the conditions of use recommended and the effectiveness of the drug for the purposes recommended.
- SOR/2018-69, ss. 15, 27
C.01.013.1 Section C.01.013 does not apply in respect of a veterinary health product.
- SOR/2017-76, s. 2
Assignment and Cancellation of Drug Identification Numbers
C.01.014 (1) No manufacturer shall sell a drug in dosage form unless a drug identification number has been assigned for that drug and the assignment of the number has not been cancelled under section C.01.014.6.
(2) Subsection (1) does not apply in respect of a veterinary health product, a study drug as defined in section C.03.301 or a medicated feed as defined in subsection 1(1) of the Feeds Regulations, 2024.
- SOR/81-248, s. 2
- SOR/97-12, s. 3
- SOR/2013-179, s. 1
- SOR/2017-259, s. 3
- SOR/2018-77, s. 3
- SOR/2024-132, s. 85
C.01.014.1 (1) A manufacturer of a drug may make an application for a drug identification number for that drug.
(2) An application under subsection (1) shall be made to the Minister in writing and shall include the following information and material:
(a) the name of the manufacturer of the drug as it will appear on the label;
(b) the pharmaceutical form in which the drug is to be sold;
(c) in the case of any drug other than a drug described in paragraph (d), the recommended route of administration;
(d) in the case of a drug for disinfection in premises, the types of premises for which its use is recommended;
(e) a quantitative list of the medicinal ingredients contained in the drug by their proper names or, if they have no proper names, by their common names;
(f) the brand name under which the drug is to be sold;
(g) an indication of whether the drug is for human use or veterinary use;
(h) the name and quantity of each colouring ingredient that is not a medicinal ingredient;
(i) the use or purpose for which the drug is recommended;
(j) the recommended dosage of the drug;
(k) the address of the manufacturer referred to in paragraph (a) and, where the address is outside the country, the name and address of the importer of the drug;
(l) the name and address of any individual, firm, partnership or corporation, other than the names and addresses referred to in paragraphs (a) and (k), that will appear on the label of the drug;
(m) in the case of a drug for veterinary use, the written text of every label to be used in connection with the drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the drug;
(m.1) in the case of a drug for human use, mock-ups of every label to be used in connection with the drug — including any package insert and any document that is provided on request and that sets out supplementary information on the use of the drug — and mock-ups of the drug’s packages;
(n) the name and title of the person who signed the application and the date of signature; and
(o) in the case of a drug for human use, an assessment as to whether there is a likelihood that the drug will be mistaken for another drug for which a drug identification number has been assigned due to a resemblance between the brand name that is proposed to be used in respect of the drug and the brand name, common name or proper name of the other drug.
(3) In the case of a new drug, a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission or an abbreviated extraordinary use new drug submission filed under section C.08.002, C.08.002.01 or C.08.002.1 shall be regarded as an application for a drug identification number.
- SOR/81-248, s. 2
- SOR/93-202, s. 4
- SOR/98-423, s. 3
- SOR/2011-88, s. 2
- SOR/2014-158, s. 7
- SOR/2017-259, s. 4
- SOR/2018-69, ss. 27, 33(F)
C.01.014.2 (1) Subject to subsection (2), if a manufacturer has provided all the information and material described in subsection C.01.014.1(2) or section C.08.002, C.08.002.01 or C.08.002.1, as the case may be, in respect of a drug, the Minister shall issue to the manufacturer a document that
(a) sets out
(i) the drug identification number assigned for the drug, preceded by the abbreviation “DIN”, or
(ii) if there are two or more brand names for the drug, the drug identification numbers assigned by the Minister for the drug, each of which pertains to one of the brand names and is preceded by the abbreviation “DIN”; and
(b) contains the information referred to in paragraphs C.01.014.1(2)(a) to (f).
(2) The Minister may refuse to issue the document referred to in subsection (1) if he or she has reasonable grounds to believe that the product in respect of which an application referred to in section C.01.014.1 has been made
(a) is not a drug, or
(b) is a drug but its sale would cause injury to the health of the purchaser or consumer or would be a contravention of the Act or these Regulations.
(3) If the Minister refuses to issue the document under subsection (2), the manufacturer may submit additional information or material and request the Minister to reconsider his or her decision.
(4) On the basis of the additional information or material submitted under subsection (3), the Minister shall reconsider the grounds on which the refusal to issue the document was made.
- SOR/81-248, s. 2
- SOR/92-230, s. 1
- SOR/98-423, s. 4
- SOR/2011-88, s. 3
- SOR/2017-259, s. 5
- SOR/2018-69, ss. 16, 27, 37
C.01.014.21 (1) The Minister may, at any time, impose terms and conditions on a drug identification number assigned for a Class B opioid or amend those terms and conditions.
(1.1) The Minister may, at any time, impose terms and conditions on a drug identification number assigned for a designated COVID-19 drug, or amend those terms and conditions, if
(a) a notice of compliance was issued under section C.08.004 in respect of
(i) a new drug submission that was filed under section C.08.002 for the designated COVID-19 drug that contains the statement referred to in paragraph C.08.002(2.1)(a), or
(ii) a supplement to a new drug submission referred to in subparagraph (i) that was filed under section C.08.003 for the designated COVID-19 drug; or
(b) a notice of compliance was issued under section C.08.004 in respect of one of the following that was filed for the designated COVID-19 drug on the basis of a direct or indirect comparison to another designated COVID-19 drug referred to in paragraph (a):
(i) a new drug submission under section C.08.002,
(ii) an abbreviated new drug submission under section C.08.002.1, or
(iii) a supplement to a new drug submission or abbreviated new drug submission under section C.08.003.
(2) The Minister shall notify, in writing, the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number of any terms and conditions imposed on the drug identification number and of any amendment to those terms and conditions.
(3) The following definitions apply in this section.
- Class B opioid
Class B opioid means a drug set out in Part B of the List of Opioids, published by the Government of Canada on its website, as amended from time to time. (opioïde de catégorie B)
- designated COVID-19 drug
designated COVID-19 drug has the same meaning as in section C.08.001.1. (drogue désignée contre la COVID-19)
- SOR/2018-77, s. 4
- SOR/2021-45, s. 2
C.01.014.3 The manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug shall, within 30 days after the day on which the drug is first sold following the issuance by the Minister of the document, date and sign the document and return it to the Minister with a statement set out on it that the information it contains is correct and with an indication of the date of that first sale.
- SOR/81-248, s. 2
- SOR/98-423, s. 5
- SOR/2014-158, s. 8
- SOR/2017-259, s. 6
- SOR/2018-69, s. 38
C.01.014.4 If the information referred to in subsection C.01.014.1(2) in respect of a drug is no longer correct owing to a change in the subject matter of the information,
(a) in the case of a change in the subject matter of any of the information referred to in paragraphs C.01.014.1(2)(a) to (f)
(i) that occurs prior to the sale of the drug, a new application shall be made, or
(ii) that occurs after the sale of the drug, no further sale of the drug shall be made until a new application for a drug identification number in respect of that drug is made and a number is assigned; and
(b) in the case of a change in the subject matter of any of the information referred to in paragraphs C.01.014.1(2)(g) to (k)
(i) that occurs prior to the sale of the drug, the particulars of the change shall be submitted with the return of the document referred to in section C.01.014.3, or
(ii) that occurs after the sale of the drug, the person to whom the drug identification number in respect of that drug was issued shall, within 30 days of the change, inform the Minister of the change.
- SOR/81-248, s. 2
- SOR/92-230, s. 2
- SOR/98-423, s. 6
- SOR/2016-139, s. 2(F)
- SOR/2018-69, s. 27
C.01.014.5 (1) The manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug shall, annually before the first day of October and in a form established by the Minister, provide the Minister with a notification that is signed by them and that
(a) indicates whether any of the following circumstances apply in respect of the drug:
(i) as of the day on which the notification is sent,
(A) the manufacturer sells the drug in Canada, or
(B) the manufacturer has discontinued the sale of the drug in Canada, or
(ii) the manufacturer has not sold the drug in Canada for a period that is greater than 12 months and a portion of that period is covered by the notification; and
(b) subject to subsection (2), confirms that the information that the manufacturer previously submitted with respect to the drug under subsection C.01.014.1(2), paragraph C.01.014.4(b) or section C.08.002, C.08.002.01, C.08.002.1 or C.08.003, as the case may be, is correct as of the day on which the notification is sent.
(2) If any of the information that the manufacturer submitted under a provision referred to in paragraph (1)(b) is not correct as of the day on which the notification is sent, the manufacturer shall update that information in the notification.
- SOR/81-248, s. 2
- SOR/2017-259, s. 7
- SOR/2018-69, ss. 27, 39
C.01.014.6 (1) The Minister shall cancel the assignment of a drug identification number for a drug if
(a) the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number advises under section C.01.014.7 that they discontinued the sale of the drug; or
(c) the Minister determines that the product for which the drug identification number has been assigned is not a drug.
(2) The Minister may cancel the assignment of a drug identification number for a drug if
(a) the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number contravenes section C.01.014.5;
(b) the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number has been notified under section C.01.013 that the evidence that they submitted with respect to the drug is not sufficient; or
(c) the drug is a new drug in respect of which the notice of compliance has been suspended under section C.08.006.
(3) The Minister may cancel the assignment of a drug identification number for a drug if, after he or she has, under section 21.31 of the Act, ordered the holder of a therapeutic product authorization referred to in subparagraph C.01.052(1)(a)(i) or (iii) to conduct an assessment of the drug in order to provide evidence establishing that the benefits associated with the drug outweigh the risks of injury to health,
(a) the holder fails to comply with the order; or
(b) the holder complies with the order but the Minister determines that the results of the assessment are not sufficient to establish that the benefits associated with the drug outweigh the risks of injury to health.
(4) For greater certainty, the Minister’s power to cancel the assignment of a drug identification number
(a) under paragraph (2)(b) is not affected by his or her power to cancel the assignment of such a number under subsection (3); and
(b) under subsection (3) is not affected by his or her power to cancel the assignment of such a number under paragraph (2)(b).
- SOR/81-248, s. 2
- SOR/2016-139, s. 3
- SOR/2017-259, s. 7
- SOR/2018-69, ss. 27, 39
- SOR/2018-84, ss. 1, 13
- SOR/2020-262, s. 1
Shortages of Drugs and Interruption and Discontinuation of Sale of Drugs
C.01.014.7 The manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug shall, within 30 days after the day on which they discontinue the sale of the drug, submit the following information to the Minister:
(a) the drug identification number assigned for the drug;
(b) the date on which the manufacturer discontinued the sale of the drug; and
(c) the latest expiration date of the drug that the manufacturer sold and the lot number of that drug.
- SOR/81-248, s. 2
- SOR/2016-139, s. 5
- SOR/2017-259, s. 8
C.01.014.71 If a period of 12 months has elapsed since the day on which the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug as defined in section C.01.014.8 last sold the drug, the manufacturer shall so notify the Minister in writing within 30 days after the day on which that period ends.
- SOR/2017-259, s. 8
C.01.014.72 If the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug resumes the sale of the drug after a period of 12 months has elapsed since the day on which they last sold the drug, the manufacturer shall so notify the Minister in writing within 30 days after the day on which they resume the sale of the drug.
- SOR/2017-259, s. 8
C.01.014.8 The following definitions apply in this section and in sections C.01.014.9 to C.01.014.14.
- drug
drug means any of the following drugs for human use for which a drug identification number has been assigned:
(a) drugs included in Schedule I, II, III, IV or V to the Controlled Drugs and Substances Act;
(b) prescription drugs;
(c) drugs that are listed in Schedule C or D to the Act; and
(d) drugs that are permitted to be sold without a prescription but that are to be administered only under the supervision of a practitioner. (drogue)
- shortage
shortage, in respect of a drug, means a situation in which the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for the drug is unable to meet the demand for the drug in Canada. (pénurie)
- SOR/2016-139, s. 5
- SOR/2017-259, s. 8
- SOR/2018-69, ss. 17, 40
- SOR/2021-199, s. 1
C.01.014.9 (1) If a shortage of a drug exists or is likely to occur, the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for the drug shall post the following information in English and French on a website that is operated by a party for that purpose with whom Her Majesty in right of Canada has entered into a contract to make that information available to the public:
(a) the manufacturer’s name and their telephone number, email address, website address, postal address or any other information that enables communication with them;
(b) the drug identification number assigned for the drug;
(c) the drug’s brand name and proper name or, if it does not have a proper name, its common name;
(d) the proper names of the drug’s medicinal ingredients or, if they do not have proper names, their common names;
(e) the drug’s therapeutic classification according to the Anatomical Therapeutic Chemical classification system (ATC), established by the World Health Organization Collaborating Centre for Drug Statistics Methodology — namely the level 3 description of, and level 4 code for, the drug;
(f) the drug’s strength;
(g) the drug’s dosage form;
(h) the quantity of the drug contained in its package;
(i) the drug’s route of administration;
(j) the date when the shortage began or is anticipated to begin;
(k) the anticipated date when the manufacturer will be able to meet the demand for the drug, if they can anticipate that date; and
(l) the actual or anticipated reason for the shortage.
(2) The manufacturer shall post the information
(a) if they anticipate that a shortage will begin in more than six months, at least six months before the day on which they anticipate it to begin;
(b) if they anticipate that a shortage will begin in six months or less, within five days after the day on which they anticipate it; or
(c) if they did not anticipate the shortage, within five days after the day on which they become aware of it.
(3) If any of the information that was posted by the manufacturer changes, they shall update that information on the website within two days after the day on which they make or become aware of the change.
(4) Within two days after the day on which the manufacturer is able to meet the demand for the drug, they shall post information on the website to that effect.
(5) This section does not apply in respect of a shortage of a drug that results from a decision by the manufacturer to discontinue its sale.
- SOR/2016-139, s. 5
- SOR/2017-259, s. 9
- SOR/2021-199, s. 2
C.01.014.10 (1) If the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug decides to discontinue the sale of the drug, they shall post the following information in English and French on the website referred to in subsection C.01.014.9(1):
(a) the manufacturer’s name and their telephone number, email address, website address, postal address or any other information that enables communication with them;
(b) the drug identification number assigned for the drug;
(c) the drug’s brand name and proper name or, if it does not have a proper name, its common name;
(d) the proper names of the drug’s medicinal ingredients or, if they do not have proper names, their common names;
(e) the drug’s therapeutic classification according to the Anatomical Therapeutic Chemical classification system (ATC), established by the World Health Organization Collaborating Centre for Drug Statistics Methodology — namely the level 3 description of, and level 4 code for, the drug;
(f) the drug’s strength;
(g) the drug’s dosage form;
(h) the quantity of the drug contained in its package;
(i) the drug’s route of administration;
(j) the date on which the manufacturer will discontinue the sale of the drug; and
(k) the reason for the discontinuation of sale.
(2) The manufacturer shall post the information
(a) if they decide to discontinue the sale of the drug in more than six months, at least six months before the day on which they will discontinue its sale; and
(b) if they decide to discontinue the sale of the drug in six months or less, within five days after the day on which that decision is made.
(3) If any of the information that was posted by the manufacturer changes, they shall update that information on the website within two days after the day on which they make or become aware of the change.
- SOR/2016-139, s. 5
- SOR/2017-259, s. 10
C.01.014.11 The Minister shall ensure that a hyperlink to the website referred to in subsection C.01.014.9(1) is on the Government of Canada website.
- SOR/2016-139, s. 5
- SOR/2017-259, s. 11
C.01.014.12 (1) The Minister may request that the manufacturer to whom a document was issued under subsection C.01.014.2(1) that sets out the drug identification number assigned for a drug — or any person who holds an establishment licence in respect of a drug — provide the Minister with information that is in their control if the Minister has reasonable grounds to believe that
(a) there is a shortage or risk of shortage of the drug;
(b) the information is necessary to establish or assess
(i) the existence of a shortage or risk of shortage of the drug,
(ii) the reason for a shortage or risk of shortage of the drug,
(iii) the effects or potential effects on human health of a shortage of the drug, or
(iv) measures that could be taken to prevent or alleviate a shortage of the drug; and
(c) the manufacturer or licensee will not provide the information without a legal obligation to do so.
(2) The manufacturer or licensee shall provide the requested information electronically in a format specified by or acceptable to the Minister within the time limit specified by the Minister.
C.01.014.13 No person who holds an establishment licence shall distribute a drug for consumption or use outside Canada unless the licensee has reasonable grounds to believe that the distribution will not cause or exacerbate a shortage of the drug.
C.01.014.14 (1) If a person who holds an establishment licence distributes a drug for consumption or use outside Canada, the licensee shall immediately create a detailed record of the information that they relied on to determine that the distribution of the drug is not prohibited by section C.01.014.13.
(2) The licensee shall retain the record for at least one year after the latest expiration date of the drug that they distributed.
Tablet Disintegration Times
C.01.015 (1) Subject to subsection (2), no person shall sell for human use a drug in the form of a tablet that is intended to be swallowed whole unless, when tested by the official method DO-25, Determination of the Disintegration Time of Tablets, dated July 5, 1989,
(a) in the case of an uncoated tablet, the tablet disintegrates in not more than 45 minutes;
(b) in the case of a plain coated tablet, the tablet disintegrates in not more than 60 minutes; and
(c) in the case where the label of the drug indicates that the tablet carries an enteric coating or a coating designed to serve a purpose similar to that of an enteric coating, the tablet does not disintegrate when exposed for 60 minutes to simulated gastric fluid, but when it is subsequently exposed for a continuous period to simulated intestinal fluid, the tablet disintegrates in not more than 60 minutes.
(2) Subsection (1) does not apply in respect of a drug in the form of a tablet where
(a) a notice of compliance in respect of the drug in the form of a tablet has been issued under section C.08.004 or C.08.004.01;
(b) [Repealed, SOR/98-423, s. 7]
(c) a dissolution or disintegration test for the drug in the form of a tablet is prescribed in Division 6 of this Part;
(d) the drug is labelled as complying with a standard contained in a publication referred to in Schedule B to the Act;
(e) the drug has been demonstrated by an acceptable method to be available to the body; or
(f) representations regarding the drug are made on its label, or in any advertisement, with respect to the site, rate or extent of release to the body of a medicinal ingredient of that drug, or the availability to the body of a medicinal ingredient of that drug.
- SOR/89-429, s. 2
- SOR/89-455, s. 3
- SOR/94-36, s. 2
- SOR/98-423, s. 7
- SOR/2011-88, s. 4
Prohibition
C.01.016 No manufacturer shall sell a drug unless the manufacturer complies with the conditions set out in sections C.01.017 to C.01.019.
- SOR/95-521, s. 2
- SOR/2011-31, s. 1
Serious Adverse Drug Reaction Reporting — Manufacturers
- SOR/2019-190, s. 1
C.01.017 The manufacturer shall submit to the Minister a report of all information relating to the following serious adverse drug reactions within 15 days after receiving or becoming aware of the information, whichever occurs first:
(a) any serious adverse drug reaction that has occurred in Canada with respect to the drug; and
(b) any serious unexpected adverse drug reaction that has occurred outside Canada with respect to the drug.
- SOR/95-521, s. 2
- SOR/2011-31, s. 1
Annual Summary Report and Case Reports
C.01.018 (1) The manufacturer shall prepare an annual summary report of all information relating to adverse drug reactions and serious adverse drug reactions to the drug that it received or became aware of during the previous 12 months.
(2) The annual summary report shall contain a concise, critical analysis of the adverse drug reactions and serious adverse drug reactions to the drug.
(3) In preparing the annual summary report, the manufacturer shall determine, on the basis of the analysis referred to in subsection (2), whether there has been a significant change in what is known about the risks and benefits of the drug during the period covered by the report and shall include its conclusions in this regard in the summary report.
(4) If, in preparing the annual summary report, the manufacturer concludes that there has been a significant change, it shall notify the Minister without delay, in writing, unless this has already been done.
(5) The Minister may, for the purposes of assessing the safety and effectiveness of the drug, request in writing that the manufacturer submit to the Minister one or both of the following:
(a) the annual summary reports;
(b) the case reports relating to the adverse drug reactions and serious adverse drug reactions to the drug that are known to the manufacturer.
(6) The Minister shall, after giving the manufacturer an opportunity to be heard, specify a period for the submission of the annual summary reports or case reports, or both, that is reasonable in the circumstances, and the manufacturer shall submit the reports within that period.
- SOR/2011-31, s. 1
C.01.018.1 Section C.01.018 does not apply in respect of a veterinary health product.
- SOR/2017-76, s. 4
Issue-Related Summary Report
C.01.019 (1) The Minister may, for the purposes of assessing the safety and effectiveness of the drug, request in writing that the manufacturer submit to the Minister an issue-related summary report.
(2) The report shall contain a concise, critical analysis of the adverse drug reactions and serious adverse drug reactions to the drug, as well as case reports of all or specified adverse drug reactions and serious adverse drug reactions to the drug that are known to the manufacturer in respect of the issue that the Minister directs the manufacturer to analyze in the report.
(3) The Minister shall, after giving the manufacturer an opportunity to be heard, specify a period for the submission of the report that is reasonable in the circumstances. The Minister may only specify a period that is less than 30 days if the Minister needs the information in the report to determine whether the drug poses a serious and imminent risk to human health.
(4) The manufacturer shall submit the report within the specified period.
- SOR/2011-31, s. 1
- SOR/2017-18, s. 14
C.01.019.1 Section C.01.019 does not apply in respect of a veterinary health product.
- SOR/2017-76, s. 5
Maintenance of Records
C.01.020 (1) The manufacturer shall maintain records of the reports and case reports referred to in sections C.01.017 to C.01.019.
(2) The manufacturer shall retain the records for 25 years after the day on which they were created.
- SOR/2011-31, s. 1
Provision of Information Under Section 21.8 of Act
C.01.020.1 (1) For the purposes of section 21.8 of the Act, hospitals are the prescribed health care institutions that shall provide information that is in their control to the Minister about a serious adverse drug reaction.
(2) The following prescribed information about a serious adverse drug reaction that is in a hospital’s control shall be provided to the Minister in writing within 30 days after the day on which the serious adverse drug reaction is first documented within the hospital:
(a) the name of the hospital and the contact information of a representative of that hospital;
(b) the drug’s brand name, proper name or common name;
(c) in the case of a drug imported under subsection C.10.001(2) or section C.10.006, the identifying code or number of the drug, if any, assigned in the country in which the drug was authorized for sale;
(c.1) in the case of a drug whose sale has been authorized under subsection C.11.003(1), its identifying name, code, number or mark;
(d) the drug identification number assigned for the drug, if applicable;
(e) the patient’s age and sex;
(f) a description of the serious adverse drug reaction;
(g) the date on which the serious adverse drug reaction was first documented;
(h) the date on which the patient first used the drug and, if applicable, the date on which the patient stopped using the drug;
(i) the date on which the serious adverse drug reaction first occurred and, if applicable, the date on which the patient’s health was restored to its state prior to the reaction;
(j) any medical condition of the patient that directly relates to the serious adverse drug reaction;
(k) any concomitant therapeutic products used by the patient; and
(l) the effect of the serious adverse drug reaction on the patient’s health.
(3) A hospital is exempt from section 21.8 of the Act in respect of the reporting of information referred to in subsection (2) if
(a) the hospital does not have in its control all of the information referred to in paragraphs (2)(b), (c), (e) and (f) in respect of the serious adverse drug reaction; or
(b) the serious adverse drug reaction relates only to any of the following drugs:
(i) a vaccine that was administered under a routine immunization program of a province,
(ii) a drug that is authorized for sale under Division 5 of this Part, or
(iii) a drug that was sold under subsection C.08.011(1).
(4) In this section, hospital means a facility
(a) that is licensed, approved or designated as a hospital by a province in accordance with the laws of the province to provide care or treatment to persons suffering from any form of disease or illness; or
(b) that is operated by the Government of Canada and that provides health services to in-patients.
Limits of Drug Dosage
C.01.021 Except as provided in these Regulations, no person shall sell a drug for human use listed in the following table unless both the inner and the outer labels other than the inner label of a single dose container carry a statement of
(a) the quantitative content of the drug,
(b) the recommended single and daily adult dose designated as such, except for
(i) preparations solely for external use, or
(ii) preparations solely for children’s use; and
(c) adequate directions for use when the drug is recommended for children which shall be either
(i) the statement “CHILDREN: As directed by the physician”, or
(ii) a suitable reduced maximum single and daily dose which shall not exceed the following:
Age in years Proportion of adult dose 10 - 14 one-half 5 - 9 one-fourth 2 - 4 one-sixth under 2 years as directed by physician TABLE
Table of Limits of Drug Dosage for Adults
Item External Use Internal Use — — Maximum Limit Maximum Dosage Unless otherwise stated, doses are in milligrams Per cent Single Daily Acetaminophen — 650 4.0 g Acetanilide and derivatives (except N-Acetyl-ρ-amino phenol) — 65 195 Acetylsalicylic Acid — 650 4.0 g Aconitine, its preparations and derivatives 0.2 0.1 0.1 Adonis vernalis — 65 195 Amylocaine, its salts and derivatives when sold or recommended for opthalmic use 0.0 0.0 0.0 Amylocaine Hydrochloride, except when sold or recommended for ophthalmic use 1.0 0.0 0.0 Antimony, compounds of — 3.3 13 Atropine, Methylatropine, and their salts 1.0 0.13 0.44 Belladonna and its preparations, on the basis of belladonna alkaloids 0.375 0.13 0.44 Benzene (Benzol) — — — Benzocaine 8.0 195 585 Beta-Naphthol — 195 585 Butacaine, its salts and derivatives when sold or recommended for ophthalmic use 0.0 0.0 0.0 Butacaine Sulphate, except when sold or recommended for opthalmic use 1.0 0.0 0.0 Cadexomer Iodine 0.0 0.0 0.0 Cantharides, cantharidin, and their preparations, on the basis of cantharidin, except blisters 0.03 0.0 0.0 Cantharides, blisters only 0.2 0.0 0.0 Cedar Oil 25.0 0.0 0.0 Chlorbutol (not more often than every 4 hours) — 325 975 Choline Salicylate — 870 5.22 g Cinchocaine Hydrochloride, except suppositories 1.0 0.0 0.0 Cinchocaine Hydrochloride, suppositories only — 11 11 Colchicine and its salts — 0.55 1.65 Colchicum and its preparations, on the basis of colchicine — 0.27 0.81 Croton Oil 10.0 0.0 0.0 Cyproheptadine and its salts — when sold or recommended for the promotion of weight gain — 0.0 0.0 Ephedrine and its salts — 11 32.5 Ephedrine and its salts, sprays 1.0 — — Epinephrine and its salts, sprays 1.0 — — Gelseminine (Gelsemine) and its salts (not to be repeated within 4 hours) — 0.55 1.65 Gelsemium and its preparations, on the basis of the crude drug — 16.2 48.6 Hydrocyanic (Prussic) Acid as 2 per cent solution — 0.062 ml 0.31 ml Hydroquinone 2.0 — — Hyoscine (Scopolamine) and its salts 0.5 0.325 0.975 Hyoscine aminoxide hydrobromide 0.5 0.325 0.975 Hyoscyamine and its salts — 0.325 0.975 Hyoscyamus and its preparations, on the basis of hyoscyamus alkaloids — 0.073 0.22 Lobelia and its preparations, on the basis of the crude drug — 130 390 Lobeline and its salts — 2.0 6.0 Magnesium Salicylate — 650 4.0 g Methyl Salicylate 30 — — Methylene Blue — 130 390 Phenacetin — 650 1.95 g Phenazone and compounds thereof — 325 975 Phenol 2.0 32.5 260 Phenylpropanolamine when sold or recommended as an appetite depressent — 0.0 0.0 Phosphorus — 0.0 0.0 Podophyllin 0.0 0.0 0.0 Potassium Chlorate — 325 975 Potassium Chlorate, gargle 2.5 — — Procaine and its salts — — — Proxymetacaine, its salts and derivatives when sold or recommended for ophthalmic use 0.0 0.0 0.0 Salicylamide — 975 2.925 g Santonin — 65 130 Selenium and its compounds 2.5 0.0 0.0 Sodium Chlorate — 325 975 Sodium Fluoride — 0.1 0.1 Sodium Salicylate — 650 4.0 g Squill and its preparations, on the basis of crude drug — 32.5 97.5 Stramonium and its preparations, on the basis of stramonium alkaloids — 0.16 0.65 Strychnine and its salts — 0.0 0.0 Tannic Acid — 150 1 000 Tetracaine, its salts and derivatives when sold or recommended for ophthalmic use 0.0 0.0 0.0 Thiocyanates 0.0 0.0 0.0 Urethane 0.0 0.0 0.0 Where drugs having similar physiological actions occur in combination, the dosage of each shall be proportionately reduced.
Accurate dosagesmay be expressed in either metric units or imperial units. If the dosage is expressed in both systems, then an approximation may be used for one expression, but such approximation must precede or follow the accurate statement by which the product will be judged and must be in brackets.
- SOR/78-422, s. 1
- SOR/80-544, s. 3
- SOR/84-145, s. 1
- SOR/85-715, s. 3
- SOR/85-966, s. 2
- SOR/88-94, s. 1
- SOR/89-229, s. 2
- SOR/89-548, s. 1
C.01.022 Notwithstanding paragraph C.01.021(b), the recommended single and daily dosage of a drug
(a) intended to be burned and the smoke inhaled may be increased to 10 times the oral dose, and
(b) intended for use as suppositories may be increased to 33 1/3 per cent in excess of the oral dose.
C.01.024 (1) Sections C.01.021 and C.01.022 do not apply to
(a) a drug sold to a drug manufacturer; or
(b) a drug sold on prescription.
(2) Paragraph C.01.021(c) does not apply to
(a) acetaminophen;
(b) acetylsalicylic acid;
(c) magnesium salicylate;
(d) sodium salicylate; or
(e) choline salicylate.
(3) Where a drug mentioned in any of paragraphs (2)(a) to (d) is recommended for children, no person shall sell the drug for human use unless both the inner and the outer labels carry a statement that it is recommended
(a) that the drug be used as directed by a physician; or
(b) that the maximum doses of the drug not exceed the amounts set out in the following table and that single doses not be administered more frequently than every four hours.
TABLE
Maximum Dose
Item Column I Column II Column III Column IV Column V Column VI Column VII Age Maximum Children’s Dose (80 mg units) Acetaminophen Drops Maximum Children’s Dose (80 mg units) Maximum Children’s Dose (160 mg units) Acetaminophen Maximum Adult’s Dose (325 mg units) Maximum Single Dose (mg) Maximum Daily Dose (mg) 1 11 to under 12 years — 6 3 1.5 480 2 400 2 9 to under 11 years — 5 2.5 1.25 400 2 000 3 6 to under 9 years — 4 2 1 320 1 600 4 4 to under 6 years — 3 1.5 — 240 1 200 5 2 to under 4 years — 2 1 — 160 800 6 1 to under 2 years 1.5 or as directed by a physician — — — 120 600 7 4 months to under 1 year 1 or as directed by a physician — — — 80 400 8 0 to under 4 months 0.5 or as directed by a physician — — — 40 200 (4) Where choline salicylate is recommended for children, no person shall sell the drug for human use unless both the inner and the outer labels carry a statement that it is recommended
(a) that the drug be used as directed by physician; or
(b) that the maximum doses of the drug not exceed the amounts set out in the following table and that single doses not be administered more frequently than every four hours.
TABLE
MAXIMUM DOSE Age (Years) Adult Dosage Units (435 mg) Single Dose (mg) Maximum Daily Dose (mg) 11 to under 12 1 ½ 660 3 300 9 to under 11 1 ¼ 550 2 750 6 to under 9 1 440 2 200 4 to under 6 ¾ 330 1 650 2 to under 4 ½ 220 1 100 Under 2 As directed by physician
- SOR/84-145, s. 2
- SOR/90-587, s. 1
C.01.025 Both the inner and the outer labels of a drug that carry a recommended single or daily dosage or a statement of concentration in excess of the limits provided by section C.01.021 shall carry a caution that the product is to be used only on the advice of a physician.
C.01.026 The provisions of section C.01.025 do not apply to
(a) a drug sold on prescription, or
(b) the inner label of a single-dose container.
C.01.027 (1) Where a person advertises to the general public a drug for human use, the person shall not make any representation other than with respect to the brand name, proper name, common name, price and quantity of the drug if it
(a) contains a drug set out in the table to section C.01.021; and
(b) carries on its label
(i) a statement of the recommended single or daily adult dosage that results in a single or daily adult dosage of the drug referred to in paragraph (a) in excess of the maximum dosage set out in the table to section C.01.021 for that drug, or
(ii) a statement that shows a concentration of the drug referred to in paragraph (a) in excess of the maximum limit set out in the table to section C.01.021 for that drug.
(2) Subsection (1) does not apply to products containing
(a) acetaminophen;
(b) acetylsalicylic acid;
(c) choline salicylate;
(d) magnesium salicylate; or
(e) sodium salicylate.
(3) [Repealed, SOR/94-409, s. 1]
(4) Where a person advertises to the general public a drug for human use that contains acetylsalicylic acid, the person shall not make any representation with respect to its administration to or use by children or teenagers.
- SOR/81-358, s. 1
- SOR/84-145, s. 3
- SOR/85-715, s. 4(F)
- SOR/85-966, s. 3
- SOR/93-202, s. 5
- SOR/93-411, s. 1
- SOR/94-409, s. 1
Cautionary Statements and Child Resistant Packages
C.01.028 (1) Subject to subsection (2), the inner and outer labels of a drug that contains
(a) acetylsalicylic acid or any of its salts or derivatives, salicylic acid or a salt thereof, or salicylamide, where the drug is recommended for children, shall carry a cautionary statement to the effect that the drug should not be administered to a child under two years of age except on the advice of a physician;
(b) boric acid or sodium borate as a medicinal ingredient shall carry a cautionary statement to the effect that the drug should not be administered to a child under three years of age;
(c) hyoscine (scopolamine) or a salt thereof shall carry a cautionary statement to the effect that the drug should not be used by persons suffering from glaucoma or where it causes blurring of the vision or pressure pain within the eye;
(d) phenacetin, either singly or in combination with other drugs, shall carry the following cautionary statement:
“CAUTION: May be injurious if taken in large doses or for a long time. Do not exceed the recommended dose without consulting a physician.”; or
(e) acetylsalicylic acid for internal use shall carry a cautionary statement to the effect that the drug should not be administered to or used by children or teenagers who have chicken pox or manifest flu symptoms before a physician or pharmacist is consulted about Reye’s syndrome, which statement shall also refer to the fact that Reye’s syndrome is a rare and serious illness.
(2) Subsection (1) does not apply to a drug that is
(a) intended for parenteral use only;
(b) dispensed pursuant to a prescription; or
(c) a prescription drug or a drug that is required to be sold under a prescription by Part G, the Benzodiazepines and Other Targeted Substances Regulations or the Narcotic Control Regulations.
- SOR/86-93, s. 2
- SOR/88-323, s. 2(F)
- SOR/93-411, s. 2
- SOR/2013-122, s. 7
C.01.029 (1) Subject to subsections C.01.031.2(1) and (2), the inner and outer labels of a drug
(a) that contains
(i) salicylic acid, a salt thereof or salicylamide,
(ii) acetylsalicylic acid, or any of its salts or derivatives,
(iii) acetaminophen, or
(iv) more than five per cent alkyl salicylates,
(a.1) that contains nicotine or any of its salts and is a vaping product as defined in section 2 of the Tobacco and Vaping Products Act but that is not referred to in paragraph 2(b) or (c) or section 3 of the Regulations Excluding Certain Vaping Products Regulated Under the Food and Drugs Act from the Application of the Tobacco and Vaping Products Act, or
(b) that is in a package that contains
(i) more than the equivalent of 250 mg of elemental iron, or
(ii) more than the equivalent of 120 mg of fluoride ion, unless the drug is intended solely for use in dentists’ offices,
shall carry a cautionary statement to the effect that the drug should be kept out of the reach of children.
(2) Subject to subsections C.01.031.2(1) and (2), the inner label and outer label of a drug that is in a package shall carry a cautionary statement, to the effect that there is enough drug in the package to seriously harm a child, if the package contains
(a) more than 1.5 g of salicylic acid or the equivalent quantity of any of its salts or salicylamide,
(b) more than 2 g of acetylsalicylic acid or the equivalent quantity of any of its salts or derivatives,
(c) more than 3.2 g of acetaminophen,
(d) more than the equivalent of 250 mg of elemental iron,
(e) more than the equivalent of 120 mg of fluoride ion, unless the drug is intended solely for use in dentists’ offices, or
(f) in the case where the drug is a vaping product as defined in section 2 of the Tobacco and Vaping Products Act but is not referred to in paragraph 2(b) or (c) or section 3 of the Regulations Excluding Certain Vaping Products Regulated Under the Food and Drugs Act from the Application of the Tobacco and Vaping Products Act, 0.1 mg/ml or more of nicotine or the equivalent quantity of any of its salts.
(3) The cautionary statements required under subsections (1) and (2) shall be preceded by a prominently displayed symbol that is octagonal in shape, conspicuous in colour and on a background of a contrasting colour.
- SOR/86-93, s. 2
- SOR/87-484, s. 2
- SOR/88-323, s. 3(F)
- SOR/90-587, s. 2
- SOR/93-468, s. 1
- SOR/2018-132, s. 1
C.01.030 [Repealed, SOR/2003-196, s. 104]
C.01.031 (1) Subject to section C.01.031.2,
(a) no person shall sell a drug described in subsection C.01.029(1) unless
(i) where the drug is recommended solely for children, it is packaged in a child resistant package,
(ii) where the drug is not recommended solely for children, other than a drug referred to in subparagraph (iii), at least one of the sizes of packages available for sale is packaged in a child resistant package, and
(iii) where the drug is a vaping product referred to in paragraph C.01.029(1)(a.1), it is packaged in a child resistant package; and
(b) where a drug described in subsection C.01.029(1) is packaged in a package that is not a child resistant package, the outer label shall carry a statement that the drug is available in a child resistant package.
(2) Subsection C.01.031.2(2) and paragraph C.01.031.2(3)(a) do not apply to a drug referred to in paragraph C.01.029(1)(a.1).
- SOR/86-93, s. 2
- SOR/87-16, s. 1
- SOR/93-468, s. 2
- SOR/2018-132, s. 2
C.01.031.1 [Repealed, SOR/87-484, s. 3]
C.01.031.2 (1) Sections C.01.029 to C.01.031 do not apply to a drug that is
(a) a prescription drug or a drug that is required to be sold under a prescription by Part G, the Benzodiazepines and Other Targeted Substances Regulations or the Narcotic Control Regulations;
(b) intended for parenteral use only;
(c) in effervescent or powder form;
(d) in suppository form;
(e) intended for topical use, unless it is a liquid preparation containing more than five per cent alkyl salicylates;
(f) packaged in a non-reclosable package containing not more than two adult standard dosage units per package; or
(g) in toothpaste form.
(2) Sections C.01.029 to C.01.031 do not apply to a drug that is repackaged by a pharmacist or practitioner at the time of sale.
(3) Section C.01.031 does not apply to a drug that is
(a) sold only in containers that have roll-on or spray applicators or permanently installed wick applicators;
(b) sold for exclusive use in animals other than household pets; or
(c) intended solely for use in dentists’ offices, or packaged for hospital use only.
- SOR/86-93, s. 2
- SOR/87-484, s. 4
- SOR/88-323, s. 5(F)
- SOR/93-468, s. 3
- SOR/2013-122, s. 8
C.01.032 No person shall sell a corticosteroid drug for ophthalmic use unless
(a) the outer label or the package insert carries, as part of the directions for use, the following statements:
“Contraindications
Viral diseases of the cornea and conjunctiva;
Tuberculosis of the eye;
Fungal diseases of the eye;
Acute purulent untreated infections of the eye, which, like other diseases caused by micro-organisms, may be masked or enhanced by the presence of the steroid.
Side Effects
Extended ophthalmic use of corticosteroid drugs may cause increased intraocular pressure in certain individuals and in those diseases causing thinning of the cornea, perforation has been known to occur.”;
and
(b) the inner label carries the statements required by paragraph (a) or instructions to see the outer label or package insert for information about contraindications and side effects.
- SOR/2018-69, s. 18(F)
C.01.033 Section C.01.032 does not apply to a corticosteroid drug that is sold by a pharmacist under a prescription.
- SOR/2013-122, s. 9
C.01.034 No person shall disseminate to a practitioner promotional literature about corticosteroid drugs for ophthalmic use unless the statements required by paragraph C.01.032(a) are included in that literature.
C.01.035 Sections C.01.032 and C.01.034 do not apply to a drug sold solely for veterinary use.
Miscellaneous
C.01.036 (1) No manufacturer or importer shall sell
(a) a drug that contains phenacetin in combination with any salt or derivative of salicylic acid;
(b) a drug for human use that contains
(i) oxyphenisatin,
(ii) oxyphenisatin acetate, or
(iii) phenisatin; or
(c) a drug for human use that contains mercury or a salt or derivative thereof, unless the drug is
(i) a drug described in Schedule C or D to the Act, or
(ii) one of the following drugs, namely,
(A) an ophthalmic drug or other drug to be used in the area of the eye,
(B) a drug for nasal administration,
(C) a drug for otic administration, or
(D) a drug for parenteral administration that is packaged in a multi-dose container,
in which the mercury or the salt or derivative thereof is present as a preservative and the manufacturer or importer has submitted evidence to the Minister demonstrating that the only satisfactory way to maintain the sterility or stability of the drug is to use that preservative.
(2) For the purpose of clause (1)(c)(ii)(A), area of the eye means the area bounded by the supraorbital and infraorbital ridges and includes the eyebrows, the skin underlying the eyebrows, the eyelids, the eyelashes, the conjunctival sac of the eye, the eyeball and the soft tissue that lies below the eye and within the infraorbital ridge.
- SOR/78-423, s. 2
- SOR/86-93, s. 3
- SOR/89-229, s. 3
- SOR/2018-69, s. 27
C.01.036.1 No person shall sell, or advertise for sale, nitrous oxide to the general public.
- SOR/78-875, s. 1
C.01.037 (1) No person shall sell to the general public a drug that is recommended solely for children if the package in which the drug is sold contains
(a) more than 1.92 g of salicylamide or salicylic acid or the equivalent quantity of a salt of salicylic acid;
(b) more than 1.92 g of acetylsalicylic acid or the equivalent quantity of a salt or derivative thereof;
(c) more than 3.2 g of acetaminophen in 160 mg dosage units; or
(d) more than 1.92 g of acetaminophen in 80 mg dosage units.
(2) Subsection (1) does not apply to a drug dispensed pursuant to a prescription.
- SOR/86-93, s. 4
- SOR/87-484, s. 5
- SOR/88-323, s. 6
- SOR/90-587, s. 3
C.01.038 A drug for human use is adulterated if it contains
(a) Strychnine or any of its salts;
(b) extracts or tinctures of
(i) Strychnos nux vomica,
(ii) Strychnos Ignatii, or
(iii) a Strychnos species containing strychnine, other than those species mentioned in subparagraphs (i) and (ii);
(c) Methapyrilene or any of its salts;
(d) Echimidine or any of its salts; or
(e) any of the following plant species or extracts or tinctures thereof:
(i) Symphytum asperum,
(ii) Symphytum x uplandicum, or
(iii) any other plant species containing echimidine.
- SOR/79-512, s. 1
- SOR/88-173, s. 1
C.01.039 In vitro diagnostic products that are or contain drugs other than drugs listed in Schedule E to the Act, and drugs listed in Schedule D to the Act that are labelled for veterinary use only, are exempt from the application of this Part.
- SOR/97-12, s. 4
C.01.040 No manufacturer or importer shall sell a drug for human use that contains as an ingredient
(a) chloroform; or
(b) arsenic or any of its salts or derivatives, other than arsenic trioxide.
- SOR/89-229, s. 4
- SOR/2013-113, s. 1
C.01.040.1 No manufacturer shall use methyl salicylate as a medicinal ingredient in a drug for internal use in humans.
- SOR/78-422, s. 2
- SOR/78-801, s. 1
- SOR/81-334, s. 2(F)
- SOR/89-176, s. 1
- SOR/92-662, s. 1
Colouring Agents
C.01.040.2 (1) No manufacturer shall use a colouring agent in a drug other than a colouring agent listed in subsections (3) and (4).
(2) No person shall import for sale a drug that contains a colouring agent other than a colouring agent listed in subsections (3) and (4).
(2.1) The following definitions apply in subsections (3) to (4.1):
- C.I. (indication of the number)
C.I. (indication of the number) means the designation used to identify a colouring agent in the Colour Index published by The Society of Dyers and Colourists, as amended from time to time; (C.I. (indication du numéro))
- D & C (indication of the colour and the number)
D & C (indication of the colour and the number) means the designation used to identify, in accordance with the Code of Federal Regulations of the United States, a colouring agent that can be used in the United States in drugs and cosmetics; (D&C (indication de la couleur et du numéro))
- FD & C (indication of the colour and the number)
FD & C (indication of the colour and the number) means the designation used to identify, in accordance with the Code of Federal Regulations of the United States, a colouring agent that can be used in the United States in food, drugs and cosmetics. (FD&C (indication de la couleur et du numéro))
(3) The following colouring agents are permitted in drugs for internal and external use, namely,
(a) ACID FUCHSIN D (D & C Red No. 33; C.I. No. 17200),
- ALIZARIN CYANINE GREEN F (D & C Green No. 5; C.I. No. 61570),
- ALLURA RED AC (FD & C Red No. 40; C.I. No. 16035),
- AMARANTH (Delisted FD & C Red No. 2; C.I. No. 16185),
- ANTHOCYANIN DERIVED FROM JUICE EXPRESSED FROM FRESH EDIBLE FRUITS OR VEGETABLES,
- ß-APO-81-CAROTENAL (C.I. No. 40820),
- BRILLIANT BLUE FCF SODIUM SALT (FD & C Blue No. 1; C.I. No. 42090),
- BRILLIANT BLUE FCF AMMONIUM SALT (D & C Blue No. 4; C.I. No. 42090),
- CANTHAXANTHIN (C.I. No. 40850),
- CARAMEL,
- CARBON BLACK (C.I. No. 77266),
- CARMINE (C.I. No. 75470),
- CARMOISINE (Delisted Ext. D & C Red No. 10; C.I. No. 14720),
- ß-CAROTENE (C.I. No. 40800),
- CHLOROPHYLL (C.I. No. 75810),
- EOSIN YS ACID FORM (D & C Red No. 21; C.I. No. 45380:2),
- EOSIN YS SODIUM SALT (D & C Red No. 22; C.I. No. 45380),
- ERYTHROSINE (FD & C Red No. 3; C.I. No. 45430),
- FAST GREEN FCF (FD & C Green No. 3; C.I. No. 42053),
- FLAMING RED (D & C Red No. 36; C.I. No. 12085),
- HELINDONE PINK CN (D & C Red No. 30; C.I. No. 73360),
- INDIGO (D & C Blue No. 6; C.I. No. 73000),
- INDIGOTINE (FD & C Blue No. 2; C.I. No. 73015),
- IRON OXIDES (C.I. Nos. 77489, 77491, 77492, 77499),
- LITHOL RUBIN B SODIUM SALT (D & C Red No. 6; C.I. No. 15850),
- LITHOL RUBIN B CALCIUM SALT (D & C Red No. 7; C.I. No. 15850:1),
- PHLOXINE B ACID FORM (D & C Red No. 27; C.I. No. 45410:1),
- PHLOXINE B SODIUM SALT (D & C Red No. 28; C.I. No. 45410),
- PONCEAU 4R (C.I. No. 16255),
- PONCEAU SX (FD & C Red No. 4; C.I. No. 14700),
- QUINOLINE YELLOW WS (D & C Yellow No. 10; C.I. No. 47005),
- RIBOFLAVIN,
- SUNSET YELLOW FCF (FD & C Yellow No. 6; C.I. No. 15985),
- TARTRAZINE (FD & C Yellow No. 5; C.I. No. 19140),
- TITANIUM DIOXIDE (C.I. No. 77891);
(b) preparations made by extending any of the colouring agents listed in paragraph (a) on a substratum of
(i) alumina,
(ii) blanc fixe,
(iii) gloss white,
(iv) clay,
(v) zinc oxide,
(vi) talc,
(vii) rosin,
(viii) aluminum benzoate,
(ix) calcium carbonate, or
(x) any combination of the substances listed in subparagraphs (i) to (ix); and
(c) preparations made by extending any sodium, potassium, aluminum, barium, calcium, strontium or zirconium salt of any of the colouring agents listed in paragraph (a) on a substratum of
(i) alumina,
(ii) blanc fixe,
(iii) gloss white,
(iv) clay,
(v) zinc oxide,
(vi) talc,
(vii) rosin,
(viii) aluminum benzoate,
(ix) calcium carbonate, or
(x) any combination of the substances listed in subparagraphs (i) to (ix).
(4) The following colouring agents are permitted in drugs for external use, namely,
(a) ACID VIOLET 43 (Ext. D & C Violet No. 2; C.I. No. 60730),
- ALIZUROL PURPLE SS (D&C Violet No. 2; C.I. No. 60725),
- ANNATTO (C.I. No. 75120),
- BISMUTH OXYCHLORIDE (C.I. No. 77163),
- CHROMIUM HYDROXIDE GREEN (PIGMENT GREEN 18 (C.I. No. 77289)),
- DEEP MAROON (D&C Red No. 34; C.I. No. 15880:1),
- DIBROMOFLUORESCEIN (SOLVENT RED 72 (C.I. No. 45370:1); ORANGE No. 5 (D & C Orange No. 5)),
- FERRIC FERROCYANIDE (C.I. No. 77510),
- GUANINE (C.I. No. 75170),
- MANGANESE VIOLET (C.I. No. 77742),
- MICA (C.I. No. 77019),
- ORANGE II (D&C Orange No. 4; C.I. No. 15510),
- PYRANINE CONCENTRATED (D&C Green No. 8; C.I. No. 59040),
- QUINIZARIN GREEN SS (D&C Green No. 6; C.I. No. 61565),
- TONEY RED (D&C Red No. 17; C.I. No. 26100),
- URANINE ACID FORM (D&C Yellow No. 7; C.I. No. 45350:1),
- URANINE SODIUM SALT (D&C Yellow No. 8; C.I. No. 45350);
- ZINC OXIDE (C.I. No. 77947);
(b) preparations made by extending any of the colouring agents listed in paragraph (a) on a substratum of
(i) alumina,
(ii) blanc fixe,
(iii) gloss white,
(iv) clay,
(v) zinc oxide,
(vi) talc,
(vii) rosin,
(viii) aluminum benzoate,
(ix) calcium carbonate, or
(x) any combination of the substances listed in subparagraphs (i) to (ix); and
(c) preparations made by extending any sodium, potassium, aluminum, barium, calcium, strontium or zirconium salt of any of the colouring agents listed in paragraph (a) on a substratum of
(i) alumina,
(ii) blanc fixe,
(iii) gloss white,
(iv) clay,
(v) zinc oxide,
(vi) talc,
(vii) rosin,
(viii) aluminum benzoate,
(ix) calcium carbonate, or
(x) any combination of the substances listed in subparagraphs (i) to (ix).
(4.1) Despite subsection (1), no manufacturer shall use a preparation made by extending any of the following substances on a substratum of mica in a drug unless the requirements in subsection (4.3) are met:
(a) titanium dioxide (C.I. No. 77891);
(b) iron oxides (C.I. Nos. 77489, 77491, 77492, 77499); or
(c) a mixed oxide made from substances referred to in both paragraphs (a) and (b).
(4.2) Despite subsection (2), no person shall import for sale a drug that contains a preparation made by extending any of the substances referred to in paragraphs (4.1)(a) to (c) on a substratum of mica unless the requirements in subsection (4.3) are met.
(4.3) The requirements referred to in subsections (4.1) and (4.2) are the following:
(a) the drug shall be in a solid dosage form intended for oral administration or in a liquid dosage form intended for oral administration or it shall be a drug intended for external use;
(b) in the case where the drug is in a solid dosage form intended for oral administration, the drug shall not contain more than 3% of the preparation;
(c) in the case where the preparation was made using iron oxides, the preparation shall not contain more than 55% iron oxides.
(5) Subsections (1), (2), (4.1) and (4.2) do not apply in respect of a drug that is represented as being solely for use in the disinfection, for disease prevention, of
(a) medical devices;
(b) health care facilities; or
(c) premises in which food is manufactured, prepared or kept.
- SOR/84-949, s. 1
- SOR/86-590, s. 1(E)
- SOR/94-460, s. 1
- SOR/95-431, s. 1
- SOR/2002-369, s. 1
- SOR/2005-95, s. 1
- SOR/2018-248, s. 1
Prescription Drugs
C.01.040.3 In deciding whether to amend the Prescription Drug List in respect of a drug, including by adding the drug to it or removing the drug from it, the Minister shall consider whether any of the following criteria apply with respect to the drug:
(a) supervision by a practitioner is necessary
(i) for the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in respect of which the drug is recommended for use, or
(ii) to monitor a disease, disorder or abnormal physical state, or its symptoms, in respect of which the drug is recommended for use, or to monitor the use of the drug;
(b) the level of uncertainty respecting the drug, its use or its effects justifies supervision by a practitioner; or
(c) use of the drug can cause harm to human or animal health or a risk to public health and the harm or the risk can be mitigated by a practitioner’s supervision.
- SOR/2013-122, s. 10
- SOR/2017-18, s. 21(F)
C.01.040.4 The Minister shall consult the general public on any proposal by the Minister to remove a drug from the Prescription Drug List.
- SOR/2013-122, s. 10
C.01.040.5 Sections C.01.040.3 and C.01.040.4 apply, with any modifications that the circumstances require, in respect of classes of drugs.
- SOR/2013-122, s. 10
C.01.041 (1) No person shall sell a prescription drug unless
(a) they are entitled under the laws of a province to dispense a prescription drug and they sell it in that province under a verbal or written prescription that they received; or
(b) they sell it under section C.01.043.
(2) In the case of a verbal prescription, the person referred to in paragraph (1)(a) or a pharmacy technician shall create a written record of the prescription that includes the following information:
(a) the date on which the prescription was received and, if applicable, the number of the prescription;
(b) the name and address of the person to whom the prescription was issued;
(c) the proper name, common name or brand name of the drug and its quantity;
(d) the person’s name and the name of the practitioner who issued the prescription; and
(e) the directions for use provided with the prescription, whether or not the practitioner authorized it to be refilled and, if refills are authorized, the number of authorized refills.
(3) The person referred to in paragraph (1)(a) shall retain the written prescription referred to in subsection (1) or the record referred to in subsection (2) for at least two years after the day on which the prescription is filled.
- SOR/78-424, s. 2
- SOR/80-543, s. 3
- SOR/93-202, s. 6
- SOR/93-407, s. 2
- SOR/2013-122, s. 11
C.01.041.1 Subject to paragraph C.01.041.3(2)(b), a pharmacist or pharmacy technician may transfer to another pharmacist or pharmacy technician a prescription for a prescription drug.
- SOR/78-424, s. 3
- SOR/2013-122, s. 11
C.01.041.2 (1) Before a pharmacist sells a drug under a prescription that was transferred under section C.01.041.1, the pharmacist or a pharmacy technician shall
(a) create a written record of the name and address of the pharmacist or pharmacy technician who transferred the prescription and, if applicable, the number of authorized refills remaining and the date of the last refill; and
(b) obtain a copy of the written prescription or of the written record that was created under subsection C.01.041(2), as the case may be, or, in the case of a verbal transfer, create a written record that includes the information referred to in that subsection.
(2) The pharmacist shall retain the documents referred to in subsection (1) for at least two years after the day on which the prescription was filled.
- SOR/78-424, s. 3
- SOR/2013-122, s. 11
C.01.041.3 (1) A pharmacist or a pharmacy technician who transfers a prescription under section C.01.041.1 shall indicate the date of transfer on the original of the written prescription or of the written record created under subsection C.01.041(2) or in a record kept under the name of the patient in question, as the case may be.
(2) When the pharmacist or pharmacy technician has transferred the prescription,
(a) the pharmacist shall not make any additional sales under the prescription; and
(b) the pharmacist or pharmacy technician shall not transfer the prescription to another pharmacist or pharmacy technician.
- SOR/78-424, s. 3
- SOR/2013-122, s. 11
C.01.041.4 [Repealed, SOR/2013-122, s. 11]
C.01.042 A person referred to in paragraph C.01.041(1)(a) shall not refill a prescription for a prescription drug unless authorized by the practitioner and, in the case of such an authorization, they shall not refill a prescription more than the number of times specified by the practitioner.
- SOR/78-424, s. 4
- SOR/2013-122, s. 11
C.01.042.1 A person referred to in paragraph C.01.041(1)(a) shall indicate on the original of or on the copy of the written prescription or the written record created under subsection C.01.041(2) or in a record kept under the name of the patient in question, as the case may be,
(a) the date on which the prescription was filled;
(b) the date of each refill, if applicable;
(c) the quantity of drug sold when the prescription was filled and, if applicable, for each refill; and
(d) the name of the person who sold the drug.
- SOR/2013-122, s. 11
C.01.043 (1) A person may sell a prescription drug to
(a) a drug manufacturer;
(b) a practitioner;
(c) a wholesale druggist;
(d) a pharmacist; or
(e) the Government of Canada or the government of a province, for the use of a department or agency of that government, on receipt of a written order signed by the minister responsible for the department or by the person in charge of the agency, or by their duly authorized representative.
(2) If a person sells a prescription drug under paragraph (1)(e), they shall retain the written order for the drug for a period of at least two years after the day on which the drug is sold.
- SOR/2013-122, s. 11
C.01.044 If a person advertises a prescription drug to the general public, the person shall not make any representation other than with respect to the brand name, the proper name, the common name and the price and quantity of the drug.
- SOR/78-424, s. 5
- SOR/93-202, s. 7
- SOR/93-407, s. 3
- SOR/2013-122, s. 11
C.01.045 No person, other than one of the following, shall import a prescription drug:
(a) a practitioner;
(b) a drug manufacturer;
(c) a wholesale druggist;
(d) a pharmacist; or
(e) a resident of a foreign country while a visitor in Canada.
- SOR/93-407, s. 4
- SOR/2013-122, s. 11
C.01.046 [Repealed, SOR/2013-122, s. 11]
C.01.047 [Repealed, SOR/80-543, s. 4]
Distribution of Drugs as Samples
- SOR/2020-74, s. 2
C.01.048 (1) If a practitioner or pharmacist has signed an order specifying the proper name or common name, the brand name and the quantity of a drug, other than the following, the person who receives the order may distribute or cause to be distributed the drug, in dosage form, as a sample to that practitioner or pharmacist if the drug meets the requirements of these Regulations:
(a) a narcotic as defined in the Narcotic Control Regulations;
(b) a controlled drug as defined in section G.01.001; or
(c) [Repealed, SOR/2020-74, s. 3]
(d) a prescription drug as defined in subsection 1(2) of the Cannabis Regulations.
(1.1) A person may distribute or cause to be distributed a prescription drug as a sample under subsection (1) only to a practitioner or pharmacist who is entitled, under the laws of the province in which they are practising, to prescribe or dispense that drug, as the case may be.
(2) An order referred to in subsection (1) may provide that the order be repeated at specified intervals during any period not exceeding six months.
(3) Despite subsection (1), a person may distribute or cause to be distributed a drug, in dosage form, as a sample to a practitioner or pharmacist without a signed order if that drug is not a prescription drug and is part of a class of drugs that is set out in column 1 of List D, and if all of the following conditions are met:
(a) the drug contains, as its only medicinal ingredients, one or more of those set out in column 2, each of which corresponds to that class, in the corresponding quantity set out in column 3, and the drug is consistent with the descriptive information set out in columns 4 to 6;
(b) the expiration date of the drug falls on a day that is
(i) at least 30 days after the day on which it is distributed, if the expiration date consists of a day, month and year, or
(ii) in a month that follows the month in which it is distributed, if the expiration date consists only of a month and year;
(c) the drug meets the requirements of these Regulations.
- SOR/93-202, s. 9
- SOR/97-228, s. 2
- SOR/2018-144, s. 366
- SOR/2019-171, s. 24
- SOR/2020-74, s. 3
C.01.049 A person who, under subsection C.01.048(1), receives an order for and distributes or causes to be distributed a drug as a sample shall
(a) maintain records showing
(i) the name, address and description of each person to whom the drug is distributed,
(ii) the brand name, quantity and form of the drug distributed, and
(iii) the date upon which each such distribution was made; and
(b) keep those records and all orders received for drugs in accordance with section C.01.048 for a period of not less than two years from the date upon which the distribution referred to in the records was made.
- SOR/93-202, s. 10
- SOR/2020-74, s. 4
C.01.049.1 A person may distribute or cause to be distributed a drug, in dosage form, as a sample to any consumer that is 18 years of age or older if that drug is not a prescription drug and is part of a class of drugs that is set out in column 1 of List D, and if all of the following conditions are met:
(a) the drug contains, as its only medicinal ingredients, one or more of those set out in column 2, each of which corresponds to that class, in the corresponding quantity set out in column 3, and the drug is consistent with the descriptive information set out in columns 4 to 6;
(b) the expiration date of the drug falls on a day that is
(i) at least 30 days after the day on which it is distributed, if the expiration date consists of a day, month and year, or
(ii) in a month that follows the month in which it is distributed, if the expiration date consists only of a month and year;
(c) the drug meets the requirements of these Regulations.
Information — Serious Risk of Injury to Human Health
C.01.050 (1) This section applies to a holder of one or more of the following therapeutic product authorizations:
(a) a drug identification number that has been assigned under subsection C.01.014.2(1); and
(b) a notice of compliance that has been issued under section C.08.004 or C.08.004.01.
(2) The holder of a therapeutic product authorization in respect of a drug that is part of a class of drugs set out in subsection (4) shall provide the Minister with information in respect of any serious risk of injury to human health that the holder receives or becomes aware of and that is relevant to the safety of the drug, regarding
(a) risks that have been communicated by any foreign regulatory authority that is set out in Part A of the List of Foreign Regulatory Authorities for the Purposes of Section C.01.050 of the Food and Drug Regulations, published by the Government of Canada on its website, as amended from time to time, or by any person who is authorized to manufacture or sell a drug within the jurisdiction of such an authority, and the manner of the communication;
(b) changes that have been made to the labelling of any drug and that have been communicated to or requested by any foreign regulatory authority that is set out in Part B of the list referred to in paragraph (a); and
(c) recalls, reassessments and suspensions or revocations of authorizations, including licences, in respect of any drug, that have taken place within the jurisdiction of any foreign regulatory authority that is set out in Part C of the list referred to in paragraph (a).
(3) The information shall be provided to the Minister within 72 hours after the holder receives or becomes aware of it, whichever occurs first.
(4) The classes of drugs are
(a) prescription drugs;
(b) drugs that are required to be sold under a prescription by Part G, the Benzodiazepines and Other Targeted Substances Regulations or the Narcotic Control Regulations; and
(c) drugs that are permitted to be sold without a prescription but that are to be administered only under the supervision of a practitioner.
(5) Despite subsection (2), a holder of a therapeutic product authorization who provided information in accordance with
(a) paragraph (2)(a) is not required to provide the same information again under that paragraph in the case where the holder receives or becomes aware of that information in respect of a foreign regulatory authority or person referred to in that paragraph; or
(b) paragraph (2)(b) or (c) is not required to provide the same information again under that paragraph in the case where the holder receives or becomes aware of that information in respect of a foreign regulatory authority referred to in that paragraph.
(6) In this section, foreign regulatory authority means a government agency or other entity outside Canada that has a legal right to control the manufacturing, use or sale of drugs within its jurisdiction and that may take enforcement action to ensure that drugs marketed within its jurisdiction comply with the applicable legal requirements.
- SOR/2018-84, s. 2
- SOR/2020-262, s. 2(F)
Recalls
C.01.051 Where a manufacturer who sells a drug in dosage form or a person who imports into and sells in Canada a drug in dosage form commences a recall of the drug, the manufacturer or importer shall forthwith submit to the Minister the following information:
(a) the proper name of the drug, the common name of the drug if there is no proper name, the brand name of the drug and the lot number;
(b) in the case of an imported drug, the names of the manufacturer and importer;
(c) the quantity of the drug manufactured or imported;
(d) the quantity of the drug distributed;
(e) the quantity of the drug remaining on the premises of the manufacturer or importer;
(f) the reasons for initiating the recall; and
(g) a description of any other action taken by the manufacturer or importer with respect to the recall.
- SOR/82-524, s. 2
- SOR/93-202, s. 11
- SOR/2018-69, s. 27
Assessments Ordered Under Section 21.31 of the Act
C.01.052 (1) The Minister’s power to make an order under section 21.31 of the Act in respect of a drug is subject to the following conditions:
(a) the person to whom the order is made shall be the holder of one or more of the following therapeutic product authorizations in respect of the drug:
(i) a drug identification number that has been assigned under subsection C.01.014.2(1),
(ii) an establishment licence that has been issued under subsection C.01A.008(1), and
(iii) a notice of compliance that has been issued under section C.08.004 or C.08.004.01; and
(b) the Minister shall have reasonable grounds to believe that
(i) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(i) or (iii), the benefits or risks of injury to health associated with the drug are significantly different than they were when the authorization was issued,
(ii) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(ii) who is an importer, the manner in which one or more of the following activities is conducted may present a risk of injury to health associated with the drug:
(A) importation, as defined in subsection C.01A.001(1), of the drug,
(B) fabrication or packaging/labelling, as defined in subsection C.01A.001(1), of the drug outside Canada, or
(C) testing of the drug outside Canada, and
(iii) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(ii) who is not an importer, the manner in which an activity that is authorized under the authorization is conducted may present a risk of injury to health associated with the drug.
(2) The Minister shall, after examining the results of an assessment that was ordered under section 21.31 of the Act in respect of a drug,
(a) provide the holder of the therapeutic product authorization with the results of the examination; and
(b) ensure that a summary of the results of the examination, together with a description of any steps that the Minister has taken or may take as a consequence of the examination, is published on the Government of Canada website.
- SOR/2018-84, s. 3
- SOR/2020-262, s. 3
Activities Ordered Under Section 21.32 of the Act
C.01.053 The Minister’s power to make an order under section 21.32 of the Act in respect of a drug is subject to the following conditions:
(a) the person to whom the order is made shall be the holder of one or more of the following therapeutic product authorizations in respect of the drug:
(i) a drug identification number that has been assigned under subsection C.01.014.2(1),
(ii) an establishment licence that has been issued under subsection C.01A.008(1), and
(iii) a notice of compliance that has been issued under section C.08.004 or C.08.004.01;
(b) the Minister shall have reasonable grounds to believe that
(i) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(i) or (iii), there are significant uncertainties relating to the benefits or harms associated with the drug,
(ii) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(ii) who is an importer, the manner in which one or more of the following activities is conducted has introduced significant uncertainties relating to the benefits or harms associated with the drug:
(A) importation, as defined in subsection C.01A.001(1), of the drug,
(B) fabrication or packaging/labelling, as defined in subsection C.01A.001(1), of the drug outside Canada, or
(C) testing of the drug outside Canada,
(iii) in the case of a holder of a therapeutic product authorization referred to in subparagraph (a)(ii) who is not an importer, the manner in which an activity that is authorized under the authorization is conducted has introduced significant uncertainties relating to the benefits or harms associated with the drug,
(iv) the holder of the therapeutic product authorization is unable to provide the Minister with information that is sufficient to manage those uncertainties, and
(v) the applicable requirements of these Regulations, together with any terms and conditions that have been imposed on the authorization, do not allow for sufficient information to be obtained to manage those uncertainties; and
(c) the Minister shall take into account the following matters:
(i) whether the activities that the holder of the therapeutic product authorization will be ordered to undertake are feasible, and
(ii) whether there are less burdensome ways of obtaining additional information about the drug’s effects on health or safety.
- SOR/2018-84, s. 3
- SOR/2020-262, s. 4
C.01.055 and C.01.056 [Repealed, SOR/82-524, s. 2]
Limits of Variability
C.01.061 (1) Where the net amount of a drug in a package is not expressed on the label in terms of number of dosage units, any 10 packages of the drug selected as provided by official method DO-31, Determination of Net Contents, dated December 7, 1988, shall contain an amount of the drug such that, when determined by that official method, the average of the net amounts of the drug in the 10 packages is not less than the net amount of the drug shown on the label.
(2) Where the net amount of a drug in a package is expressed on the label in terms of the number of dosage units, any 10 packages of the drug selected as provided by official method DO-31, Determination of Net Contents, dated December 7, 1988, shall contain a number of units such that, when determined by that official method,
(a) the average number of dosage units in the 10 packages is not less than the number of dosage units shown on the label;
(b) no package contains less than the number of dosage units shown on the label except as provided in the table; and
(c) where the drug is a controlled drug as defined in section G.01.001 or a narcotic as defined in the Narcotic Control Regulations, no package contains more than the number of dosage units shown on the label except as provided in the table to this section.
TABLE
Item Column I Column II Labelled Number of Dosage Units Per Package Permitted Variation from the Labelled Number 1 50 or less 0 2 More than 50, but less than 101 1 3 101 or more the greater of one unit or 0.75% of the labelled number, rounded up to the next whole number
- SOR/82-429, s. 4
- SOR/89-455, s. 4
- SOR/97-228, s. 3
- SOR/2019-171, s. 24
C.01.062 (1) Subject to subsections (2) to (5), no manufacturer shall sell a drug in dosage form where the amount of any medicinal ingredient therein, determined using an acceptable method, is
(a) less than 90.0 per cent of the amount of the medicinal ingredient shown on the label; or
(b) more than 110.0 per cent of the amount of the medicinal ingredient shown on the label.
(2) Subject to subsection (5), where a drug in dosage form contains a medicinal ingredient that is a volatile substance of botanical origin or its synthetic equivalent, the amount of that ingredient, determined using an acceptable method, shall be
(a) not less than 85.0 per cent of the amount of the medicinal ingredient shown on the label; and
(b) not more than 120.0 per cent of the amount of the medicinal ingredient shown on the label.
(3) Subject to subsection (5), where a drug in capsule form contains a medicinal ingredient that is a vitamin in a fish-liver oil, no variation from the amount of the medicinal ingredient as shown on the label, determined using an acceptable method, is permitted other than that which is in accordance with the variation for that fish-liver oil as stated in any publication whose name is referred to in Schedule B to the Act.
(4) Subject to subsection (5), where a drug in dosage form contains a medicinal ingredient that is a vitamin, no variation from the amount of the medicinal ingredient shown on the label, determined using an acceptable method, is permitted other than the variation set out in column III or IV of an item of the table to this section opposite the vitamin set out in column I of that item for the amount of vitamin set out in column II of that item.
(5) Subsections (1) to (4) do not apply in respect of
(a) a drug for which a notice of compliance has been issued under section C.08.004 or C.08.004.01;
(b) [Repealed, SOR/98-423, s. 8]
(c) a drug for which a standard is contained in any publication whose name is referred to in Schedule B to the Act;
(d) a drug described in Schedule C or D to the Act or Division 6 of Part C of these Regulations; or
(e) a drug for which a drug identification number has been assigned under subsection C.01.014.2(1) and in respect of which
(i) the conditions of pharmaceutical production and quality control are suitable for controlling the identity, quality, purity, stability, safety, strength and potency of the drug,
(ii) all labels to be used in connection with the drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the drug, make proper claims in respect of the drug,
(iii) the drug can, without undue foreseeable risk to humans, be used for the purposes and under the conditions of use recommended by the manufacturer, and
(iv) the drug is effective for the purposes and under the conditions of use recommended by the manufacturer.
TABLE
Item Column I Column II Column III Column IV Vitamin Recommended daily dose Limits of variation when the recommended daily dose shown on label is equal to or less then amount set out in column II Limits of variation when the recommended daily dose shown on label is greater than amount set out in column II 1 vitamin A (or as B-carotene) 10 000 I.U. 90.0 - 165.0 % 90.0 - 115.0 % 2 thiamine 4.5 mg 90.0 - 145.0 % 90.0 - 125.0 % 3 riboflavin 7.5 mg 90.0 - 125.0 % 90.0 - 125.0 % 4 niacin or niacinamide 45 mg 90.0 - 125.0 % 90.0 - 125.0 % 5 pyridoxine 3 mg 90.0 - 125.0 % 90.0 - 125.0 % 6 d-pantothenic acid 15 mg 90.0 - 135.0 % 90.0 - 125.0 % 7 folic acid 0.4 mg 90.0 - 135.0 % 90.0 - 115.0 % 8 vitamin B12 14 µg 90.0 - 135.0 % 90.0 - 125.0 % 9 vitamin C 150 I.U. 90.0 - 145.0 % 90.0 - 125.0 % 10 vitamin D 400 I.U. 90.0 - 145.0 % 90.0 - 115.0 % 11 vitamin E 25 I.U. 90.0 - 125.0 % 90.0 - 125.0 % 12 vitamin K 0.0 mg 90.0 - 115.0 % 13 biotin 0.0 mg 90.0 - 135.0 %
- SOR/92-131, s. 1
- SOR/92-591, s. 2
- SOR/94-689, s. 2(E)
- SOR/95-530, s. 2
- SOR/98-423, s. 8
- SOR/2011-88, s. 5
- SOR/2014-158, s. 9
- SOR/2018-69, s. 19(F)
C.01.063 [Repealed, SOR/96-399, s. 2]
C.01.064 Where a drug is prepared for ophthalmic or parenteral use and contains a preservative ingredient, that ingredient
(a) shall be present only in an amount necessary to obtain the intended action and that does not pose undue risk to humans or animals; and
(b) shall not interfere with the therapeutic properties of the drug.
- SOR/90-586, s. 2
C.01.065 No person shall sell a drug that is prepared for ophthalmic or parenteral use unless a representative sample of each lot of the drug in its immediate container
(a) is tested by an acceptable method for identity, and the drug is found to be true to its proper name, or to its common name if there is no proper name;
(b) is tested by an acceptable method for sterility, except
(i) for living vaccines, or
(ii) where the manufacturer has submitted evidence, satisfactory to the Minister to prove that processing controls ensure the sterility of the drug in its immediate container,
and the drug is found to be sterile; and
(c) is subjected to such further tests satisfactory to the Minister to ensure that the drug is safe to use according to directions.
- SOR/86-552, s. 1
- SOR/90-586, s. 3
- SOR/93-202, s. 12
- SOR/96-399, s. 3
- SOR/2018-69, s. 27
C.01.066 No person shall sell a drug in aqueous solution that is prepared for parenteral use unless it has been prepared with non-pyrogenic water produced by distillation or reverse osmosis.
C.01.067 (1) Subject to subsection (2), no person shall sell a drug that is prepared for parenteral use unless a representative sample of each lot of the drug in its immediate container
(a) is tested by an acceptable method for the presence of pyrogens; and
(b) when so tested, is found to be non-pyrogenic.
(2) Subsection (1) does not apply in respect of a drug that cannot be tested for the presence of pyrogens or that is inherently pyrogenic.
- SOR/81-335, s. 1
- SOR/96-399, s. 4
C.01.068 Detailed records of the tests required by sections C.01.065 and C.01.067 shall be retained by the manufacturer for a period of at least one year after the expiration date on the label of the drug.
- SOR/85-715, s. 5
- SOR/92-654, s. 3
C.01.069 The packaging of a drug that is prepared for parenteral use shall meet the following requirements:
(a) the immediate container shall be of such material and construction that
(i) no deleterious substance is yielded to the drug,
(ii) it is non-reactive with the drug,
(iii) visual or electronic inspection of the drug is possible,
(iv) protection against environmental factors that cause deterioration or contamination of the drug is provided or, where that protection cannot be provided by the immediate container, it is provided by the secondary packaging, and
(v) a sufficient quantity of the drug is contained to allow withdrawal of the labelled amount of the drug; and
(b) the immediate closures and any material coming into contact with the drug in its immediate container shall meet the requirements of subparagraphs (a)(i) and (ii).
- SOR/96-399, s. 5
C.01.070 No person shall sell a drug that is a hypodermic tablet that does not completely dissolve in and form a clear solution with water.
Mercuric Chloride Tablets
C.01.071 No person shall sell mercuric chloride tablets for household use that are packaged in lots of 200 or less, unless
(a) such tablets are
(i) of an irregular or angular shape,
(ii) coloured blue, and
(iii) packed in an immediate container that is readily distinguishable by touch; and
(b) the principal display panel of both the inner and the outer labels carries in prominent type and in a colour contrasting to that of such labels
(i) the design of a skull and cross-bones, and
(ii) the word “Poison”.
- SOR/2001-181, s. 2
C.01.081 [Repealed, SOR/80-544, s. 4]
C.01.085 [Repealed, SOR/80-544, s. 5]
Synthetic Sweeteners
C.01.101 (1) [Repealed, SOR/78-422, s. 3]
(2) [Repealed, SOR/78-800, s. 1]
(3) [Repealed, SOR/78-422, s. 3]
C.01.121 and C.01.122 [Repealed, SOR/80-544, s. 6]
Aminopyrine and Dipyrone
C.01.131 No person shall sell Aminopyrine or Dipyrone (a derivative of Aminopyrine) for oral or parenteral use, unless
(a) the inner label carries the statement:
“WARNING: Fatal agranulocytosis may be associated with the use of Aminopyrine and Dipyrone. It is essential that adequate blood studies be made. (See enclosed warnings and precautions)”; and
(b) the outer label or the package insert carries the following statements:
“WARNING: Serious and even fatal agranulocytosis is known to occur after the administration of Aminopyrine or Dipyrone. Fatal agranulocytosis has occurred after short term, intermittent and prolonged therapy with the drugs. Therefore, the use of these drugs should be as brief as possible. Bearing in mind the possibility that such reactions may occur, Aminopyrine or Dipyrone should be used only when other less potentially dangerous agents are ineffective.
PRECAUTIONS: It is essential that frequent white blood cell counts and differential counts be made during treatment with these drugs. However, it is emphasized that agranulocytosis may occur suddenly without prior warning. The drug should be discontinued at the first evidence of any alteration of the blood count or sign of agranulocytosis, and the patient should be instructed to discontinue use of the drug at the first indication of sore throat or sign of other infection in the mouth or throat (pain, swelling, tenderness, ulceration).”
- SOR/2018-69, s. 34(F)
C.01.132 No person shall disseminate to a practitioner promotional literature about Aminopyrine or Dipyrone unless the statements set out in section C.01.131 are included in such literature.
C.01.133 The provisions of sections C.01.131 and C.01.132 do not apply to preparations containing Aminopyrine or Dipyrone that are
(a) dispensed by a pharmacist pursuant to a prescription; or
(b) sold for veterinary use only.
Coated Potassium Salts
C.01.134 No person shall sell coated tablets containing potassium salts, with or without thiazide diuretics, unless the inner label thereof or the package insert carries the following statement:
“WARNING: A probable association exists between the use of coated tablets containing potassium salts, with or without thiazide diuretics, and the incidence of serious small bowel ulceration. Such preparations should be used only when adequate dietary supplementation is not practical, and should be discontinued if abdominal pain, distension, nausea, vomiting or gastro-intestinal bleeding occur.”
- SOR/2018-69, s. 34(F)
C.01.135 No person shall disseminate to a practitioner promotional literature about coated tablets containing potassium salts, with or without thiazide diuretics, unless the statement set out in section C.01.134 is included in such literature.
C.01.136 The provisions of sections C.01.134 and C.01.135 do not apply to coated tablets containing potassium salts with or without thiazide diuretics that
(a) are sold for veterinary use only;
(b) are dispensed by a pharmacist pursuant to a prescription; or
(c) contain 100 milligrams or less of elemental potassium per tablet.
Antibiotics
C.01.401 Except as provided in these Regulations, an antibiotic for other than parenteral use shall, in addition to meeting the requirements of section C.01.004, carry on both the inner label and outer label the potency of the drug, expressed in terms of International Units where established or, if no International Unit has been established, in terms of units, milligrams, micrograms or fractions of a gram,
(a) per gram in the case of solids or viscous liquids;
(b) per millilitre in the case of other liquids; and
(c) per individual dosage or dispensing form in the case of antibiotic preparations put up in individual dosage or dispensing form.
- SOR/80-544, s. 7
- SOR/92-654, s. 4
C.01.402 [Repealed, SOR/92-654, s. 4]
C.01.410 to C.01.412 [Repealed, SOR/80-544, s. 8]
C.01.420 to C.01.422 [Repealed, SOR/80-544, s. 8]
Chloramphenicol
C.01.430 to C.01.432 [Repealed, SOR/80-544, s. 8]
C.01.433 No person shall sell chloramphenicol and its salts and derivatives, for oral or parenteral use, unless
(a) the inner label carries a warning statement to the effect that
(i) bone marrow depression has been associated with the use of chloramphenicol, and
(ii) the enclosed warnings and precautions should be read carefully; and
(b) the outer label or the package insert carries the following:
(i) a warning statement to the effect that chloramphenicol should not be used in the treatment or prophylaxis of minor infections or where it is not indicated, as in cold, influenza, or infections of the upper respiratory tract; that there are two types of bone marrow depression associated with the use of chloramphenicol; that some degree of depression of the bone marrow is commonly seen during therapy, is dose-related and is potentially reversible; that blood studies may detect early changes and; that the other type of bone marrow depression, a sudden, delayed and usually fatal bone marrow hypoplasia that may occur without warning, is very rare, and
(ii) a statement of precautions to be taken to the effect that it is essential that appropriate blood studies be made during treatment with chloramphenicol and that while blood studies may detect early peripheral blood changes, such studies cannot be relied on to detect the rare and generally irreversible bone marrow depression prior to development of aplastic anemia.
- SOR/2018-69, s. 35(F)
C.01.434 Section C.01.433 does not apply to chloramphenicol and its salts or derivatives that are sold by a pharmacist under a prescription.
- SOR/2013-122, s. 12
C.01.435 No person shall disseminate to a practitioner promotional literature about chloramphenicol and its salts or derivatives for oral or parenteral use unless the statements set out in paragraph C.01.433(b) are included in such literature.
C.01.436 The provisions of sections C.01.433 and C.01.435 do not apply to a drug sold solely for veterinary use.
C.01.440 to C.01.442 [Repealed, SOR/80-544, s. 8]
C.01.450 to C.01.452 [Repealed, SOR/80-544, s. 8]
C.01.460 to C.01.462 [Repealed, SOR/80-544, s. 8]
C.01.470 to C.01.472 [Repealed, SOR/80-544, s. 8]
C.01.480 [Repealed, SOR/80-544, s. 8]
C.01.490 to C.01.497 [Repealed, SOR/80-544, s. 8]
C.01.510 to C.01.513 [Repealed, SOR/80-544, s. 8]
C.01.520 to C.01.522 [Repealed, SOR/80-544, s. 8]
C.01.530 to C.01.532 [Repealed, SOR/80-544, s. 8]
C.01.540 to C.01.542 [Repealed, SOR/80-544, s. 8]
C.01.550 to C.01.552 [Repealed, SOR/80-544, s. 8]
C.01.560 to C.01.563 [Repealed, SOR/80-544, s. 8]
C.01.570 to C.01.572 [Repealed, SOR/80-544, s. 8]
C.01.580 [Repealed, SOR/80-544, s. 8]
C.01.590 to C.01.592 [Repealed, SOR/80-544, s. 8]
Veterinary Drugs
C.01.600 No person shall sell for veterinary use a drug listed in the Table of Limits of Drug Dosage for Adults, other than a drug in a form not suitable for human use, unless both the inner and outer labels carry the statement “For Veterinary Use Only” or “Veterinary Use Only”.
- SOR/80-543, s. 5
C.01.601 [Repealed, SOR/93-407, s. 6]
C.01.602 The provisions of sections C.01.401 and C.01.402 do not apply to an antibiotic in amounts less than 50 parts per million contained in an animal food.
C.01.603 The provisions of paragraphs C.01.401 (b) and (c) and section C.01.402 do not apply to an antibiotic in amounts greater than 50 parts per million contained in an animal food.
C.01.604 Both the inner and outer labels of a veterinary drug represented as containing a vitamin shall carry
(a) a statement of the amount of each vitamin present in the drug, expressed in terms of the proper name only of the vitamin in
(i) International Units per gram or per millilitre for vitamin A, provitamin A, vitamin D, and vitamin E,
(ii) milligrams per gram in the case of solids or viscous liquids, or per millilitre in the case of other liquids, for thiamine, riboflavin, niacin, niacinamide, pyridoxine, d-pantothenic acid, d-panthenol, folic acid, ascorbic acid, and vitamin K,
(iii) micrograms per gram in the case of solids or viscous liquids, or per millilitre in the case of other liquids, for biotin, and vitamin B12,
(iv) Oral Units for vitamin B12 with intrinsic factor concentrate, or
(v) for vitamin products put up in individual dosage or dispensing form, the specified units per individual dosage or dispensing form;
(b) except for drugs in a form not suitable for human use, the statement “For Veterinary Use Only” or “Veterinary Use Only”.
- SOR/80-543, s. 6
C.01.605 An antibiotic for parenteral use that is recommended for veterinary use only shall carry on both the inner and outer labels
(a) the potency of the drug expressed in terms of International Units where established, or, if no International Unit has been established, in terms of units, milligrams or fractions of a gram, per gram in the case of solids or viscous liquids, per millilitre in the case of other liquids, or per individual dosage or dispensing form for antibiotic preparations put up in individual dosage or dispensing form; and
(b) [Repealed, SOR/92-654, s. 5]
(c) the statement “For Veterinary Use Only” or “Veterinary Use Only”.
- SOR/80-543, s. 7
- SOR/92-654, s. 5
C.01.606 No person shall sell an antibiotic preparation for the treatment of animals, other than an antibiotic preparation that is a new drug sold pursuant to section C.08.013, unless,
(a) where the preparation is not to be used for lactating animals providing milk to be consumed as food, the inner and outer labels of the preparation carry a statement to that effect; or
(b) where the preparation may be used for lactating animals providing milk to be consumed as food,
(i) there has been submitted, on request, to the Minister, acceptable evidence to show the period of time, not exceeding 96 hours, that must elapse after the last treatment with the preparation in order that the milk from treated lactating animals will contain no residue of antibiotics that would cause injury to human health, and
(ii) the principal display panel of the outer label of the preparation, the inner label and the packaging insert, if any, describing the antibiotic preparation carry the warning “WARNING: MILK TAKEN FROM TREATED ANIMALS DURING TREATMENT AND WITHIN ... HOURS AFTER THE LATEST TREATMENT MUST NOT BE USED AS FOOD”, where the number of hours to be inserted is determined according to evidence submitted pursuant to subparagraph (i).
- SOR/88-378, s. 1
- SOR/92-664, s. 2
- SOR/93-467, s. 1
- SOR/2018-69, s. 27
C.01.606.1 No person shall sell a product intended for the prevention or treatment of foot rot of cattle if that product contains Ethylenediamine Dihydroiodide (EDDI).
- SOR/90-327, s. 1
C.01.607 Notwithstanding subparagraph C.01.004(1)(c)(ii), the declaration of a lot number is not required on the label of an animal feeding-stuff containing a drug.
- SOR/80-543, s. 8
C.01.608 The provisions of section C.01.604 do not apply to medicated feeds registered under the Feeds Act.
C.01.609 Despite paragraph C.01.401(a), the potency of an antibiotic in amounts greater than 50 parts per million contained in a medicated feed registered under the Feeds Act may be declared in grams per tonne.
- SOR/2018-69, s. 20
- SOR/2021-46, s. 6(F)
C.01.610 No person shall sell any substance having oestrogenic activity for administration to poultry that may be consumed as food.
C.01.610.1 No person shall sell a drug for administration to animals that produce food or that are intended for consumption as food if that drug contains
(a) chloramphenicol or its salts or derivatives;
(b) a 5-nitrofuran compound;
(c) clenbuterol or its salts or derivatives;
(d) a 5-nitroimidazole compound; or
(e) diethylstilbestrol or other stilbene compounds.
- SOR/85-539, s. 1
- SOR/85-685, s. 2
- SOR/91-546, s. 1
- SOR/94-568, s. 2
- SOR/97-510, s. 2
- SOR/2003-292, s. 3
C.01.610.2 No person shall sell an antibiotic preparation containing chloramphenicol, its salts or derivatives, for administration to animals that do not produce food and that are not intended for consumption as food unless
(a) both the inner label and outer label of the preparation carry the words “WARNING: FEDERAL LAW PROHIBITS THE ADMINISTRATION OF THIS PREPARATION TO ANIMALS THAT PRODUCE FOOD OR ANIMALS THAT ARE INTENDED FOR CONSUMPTION AS FOOD / MISE EN GARDE : EN VERTU DES LOIS FÉDÉRALES, IL EST INTERDIT D’ADMINISTRER CETTE PRÉPARATION AUX ANIMAUX QUI PRODUISENT DES ALIMENTS OU AUX ANIMAUX DESTINÉS À ÊTRE CONSOMMÉS COMME ALIMENTS”;
(b) where the preparation is for parenteral use, the preparation contains, in the form of chloramphenicol sodium succinate, not more than one gram of chloramphenicol per vial;
(c) where the preparation is for ophthalmic use, the preparation contains not more than one per cent chloramphenicol; and
(d) where the preparation is for oral use, the preparation
(i) is in tablet or capsule form and contains not more than one gram of chloramphenicol per tablet or capsule, or
(ii) is in the form of a chloramphenicol palmitate suspension and contains not more than three grams of chloramphenicol per container.
- SOR/91-546, s. 1
C.01.611 (1) The Minister may, in writing, from time to time require the manufacturer of a drug recommended for administration to animals that may be consumed as food
(a) to file with the Minister in respect of that drug a submission describing in detail tests carried out to verify that the administration of the drug to an animal does not result in a substance named in column II of the List referred to in the Marketing Authorization for Maximum Residue Limits for Veterinary Drugs in Foods being present in a food set out in column III of the List, except in an amount within the maximum residue limit set out in column IV of the List in respect of the food and the substance; and
(b) to print on the principal display panel of the outer label, the inner label and the packaging insert, if any, that describes the drug, a warning that food derived from animals to which the drug has been administered must not be sold for human consumption unless there has elapsed since the administration of the drug a period of time specified by the Minister, based on a review of the available data with respect to drug residues.
(2) No manufacturer shall sell a drug in respect of which the Minister has required a warning to be printed pursuant to paragraph (1)(b) unless the manufacturer has complied with that request.
- SOR/93-467, s. 2
- SOR/2016-74, s. 10
- SOR/2018-69, s. 27
C.01.612 (1) Every manufacturer or importer who sells a veterinary drug in dosage form that contains an active pharmaceutical ingredient that is set out in List A, or every person who compounds such a drug, shall, in a form established by the Minister, submit to the Minister an annual report identifying for each drug, the total quantity sold or compounded and an estimate of the quantity sold or compounded for each intended animal species.
(2) The annual report described in subsection (1) is for a period of one calendar year and shall be submitted on or before March 31 of the year following the calendar year covered by the report, beginning with the first full calendar year after the day on which this section comes into force.
- SOR/2017-76, s. 6
C.01.613 (1) No person shall import a drug into Canada for the purpose of administering it to an animal that produces food or an animal that is intended for consumption as food if the sale of the drug in Canada would constitute a violation of the Act or these Regulations.
(2) Subsection (1) does not apply to a drug that is described in List B.
- SOR/2017-76, s. 6
C.01.614 (1) Sections 43 to 58 of the Natural Health Products Regulations apply in relation to a veterinary health product, as if that product were a natural health product as defined in subsection 1(1) of those Regulations.
(2) A veterinary health product shall display, on the principal display panel of the inner and outer label, the statement: “Veterinary Health Product / Produit de santé animale” or “Produit de santé animale / Veterinary Health Product”.
(3) Section C.01.600 and paragraph C.01.604(b) do not apply in respect of a veterinary health product.
- SOR/2017-76, s. 6
C.01.615 (1) Every manufacturer or importer of a veterinary health product shall notify the Minister of the sale of that product in Canada at least 30 days before the day on which that sale is commenced.
(2) The notification shall be in a form established by the Minister and contain the following information:
(a) the name, mailing address, telephone number and email address of the manufacturer or importer;
(b) the brand name under which the veterinary health product is sold;
(c) the pharmaceutical form in which the veterinary health product is sold;
(d) the strength per dosage unit;
(e) the route of administration;
(f) a quantitative list of the medicinal ingredients and a qualitative list of the non-medicinal ingredients;
(g) the species of animal for which the veterinary health product is recommended; and
(h) the use or purpose for which the veterinary health product is recommended.
(3) A manufacturer or importer who has provided the Minister with a notification under subsection (1) shall provide the Minister with any changes to the information required under subsection (2), in a form established by the Minister, at least 30 days before the day on which the veterinary health product to which the changes relate is sold.
- SOR/2017-76, s. 6
C.01.616 If the Minister has reasonable grounds to believe that a veterinary health product may no longer be safe, the Minister may request that the manufacturer or importer of the veterinary health product provide the Minister, within 15 days after the day on which the request is received, with information and documents demonstrating that the veterinary health product is safe.
- SOR/2017-76, s. 6
C.01.617 (1) The Minister may direct the manufacturer or importer to stop the sale of a veterinary health product if
(a) the manufacturer or importer does not, within the required period, provide the Minister with the information and documents requested under section C.01.616;
(b) the information and documents provided by the manufacturer or importer in accordance with section C.01.616 do not demonstrate that the veterinary health product is safe; or
(c) the Minister has reasonable grounds to believe that the sale of the veterinary health product would be a violation of the Act or these Regulations.
(2) The Minister shall lift a direction to stop the sale of a veterinary health product if the manufacturer or importer provides the Minister with information and documents demonstrating that
(a) in the case of a direction to stop a sale under either paragraph (1)(a) or (b), the veterinary health product is safe;
(b) in the case of a direction to stop a sale under paragraph 1(c), the sale of the veterinary health product would no longer be a violation of the Act or these Regulations; or
(c) the situation giving rise to the direction to stop the sale of the veterinary health product did not exist.
- SOR/2017-76, s. 6
Contraceptive Drugs
C.01.625 Contraceptive drugs that are manufactured, sold or represented for use in the prevention of conception and that are not prescription drugs may be advertised to the general public.
- SOR/2013-122, s. 13
DIVISION 1AEstablishment Licences
Interpretation
C.01A.001 (1) The definitions in this subsection apply in this Division and in Divisions 2 to 4.
- active ingredient
active ingredient means a drug that, when used as a raw material in the fabrication of a drug in dosage form, provides its intended effect. (ingrédient actif)
- active pharmaceutical ingredient
active pharmaceutical ingredient means an active ingredient that is used in the fabrication of a pharmaceutical. (ingrédient actif pharmaceutique)
- antimicrobial agent
antimicrobial agent means a drug that is capable of destroying pathogenic micro-organisms and that is labelled as being for use in the disinfection of environmental surfaces or medical devices, as defined in the Medical Devices Regulations, that
(a) are not invasive devices as defined in those Regulations; and
(b) are intended to come into contact with intact skin only. (agent antimicrobien)
- batch certificate
batch certificate means a certificate issued by the fabricator of a lot or batch of a drug that is either imported within the framework of a mutual recognition agreement or referred to on the List of Non-prescription Drugs Not Subject to Certain Testing Requirements, and in which the fabricator
(a) identifies the master production document for the drug and certifies that the lot or batch has been fabricated, packaged/labelled and tested in accordance with the procedures described in that document;
(b) provides a detailed description of the drug, including
(i) a statement of all properties and qualities of the drug, including the identity, potency and purity of the drug, and
(ii) a statement of tolerances for the properties and qualities of the drug;
(c) identifies the analytical methods used in testing the lot or batch and provides details of the analytical results obtained;
(d) sets out the addresses of the buildings at which the lot or batch was fabricated, packaged/labelled and tested; and
(e) certifies that the lot or batch was fabricated, packaged/labelled and tested
(i) in the case of a drug that is imported within the framework of a mutual recognition agreement, in accordance with the good manufacturing practices of the regulatory authority that has recognized those buildings as meeting its good manufacturing practices standards, or
(ii) in the case of a drug that is not imported within the framework of a mutual recognition agreement and that is referred to on the List of Non-prescription Drugs Not Subject to Certain Testing Requirements, in accordance with the requirements of Division 2. (certificat de lot)
- bulk process intermediate
bulk process intermediate means an active ingredient that is used in the fabrication of either a drug of biological origin that is listed in Schedule C to the Act or a drug that is listed in Schedule D to the Act. (produit intermédiaire en vrac)
- class monograph
class monograph means a document prepared by the Department of Health that
(a) lists the types and strengths of medicinal ingredients that may be contained in drugs of a specified class; and
(b) sets out labelling and other requirements that apply to those drugs. (monographie de classe)
- dilute drug premix
dilute drug premix means a drug for veterinary use that results from mixing a drug premix with a feed, as defined in section 2 of the Feeds Act, to such a level that at least 10 kg of the resulting mixture is required to medicate one tonne of complete feed, as defined in subsection 1(1) of the Feeds Regulations, 2024, with the lowest approved dosage level of the drug. (prémélange médicamenteux dilué)
- dosage form class
dosage form class means a parenteral, tablet, capsule, solution, suspension, aerosol, powder, suppository, medical gas or drug premix, or any other dosage form class designated by the Minister. (classe de forme posologique)
- drug premix
drug premix means a drug for veterinary use to which a drug identification number has been assigned, where the directions on its label specify that it is to be mixed with feed as defined in section 2 of the Feeds Act. (prémélange médicamenteux)
- fabricate
fabricate means to prepare and preserve a drug for the purposes of sale. (manufacturer)
- import
import means to import into Canada a drug for the purpose of sale. (importer)
- List of Non-prescription Drugs Not Subject to Certain Testing Requirements
List of Non-prescription Drugs Not Subject to Certain Testing Requirements means the document entitled List of Non-prescription Drugs for Which the Testing Requirements Set Out in Subsections C.02.019(1) and (2) of the Food and Drug Regulations Do Not Apply that is published by the Government of Canada on its website, as amended from time to time. (Liste de drogues vendues sans ordonnance et non soumises à certaines analyses)
- MRA country
MRA country means a country that is a participant in a mutual recognition agreement with Canada. (pays participant)
- mutual recognition agreement
mutual recognition agreement means an international agreement that provides for the mutual recognition of compliance certification for good manufacturing practices for drugs. (accord de reconnaisance mutuelle)
- package/label
package/label means to put a drug in its immediate container or to affix the inner or outer label to the drug. (emballer-étiqueter)
- pharmaceutical
pharmaceutical means a drug other than a drug listed in Schedule C or D to the Act. (produit pharmaceutique)
- recognized building
recognized building means, in respect of the fabrication, packaging/labelling or testing of a drug, a building that a regulatory authority that is designated under subsection C.01A.019(1) in respect of that activity for that drug has recognized as meeting its good manufacturing practices standards in respect of that activity for that drug. (bâtiment reconnu)
- recognized country or region
recognized country or region means a country or region that is set out in the document entitled List of Foreign Countries or Regions and Their Regulatory Authorities for the Application of Subsection C.02.019(5) of the Food and Drug Regulations, published by the Government of Canada on its website, as amended from time to time. (pays ou régions reconnus)
- regulatory authority
regulatory authority means a government agency or other entity in an MRA country that has a legal right to control the use or sale of drugs within that country and that may take enforcement action to ensure that drugs marketed within its jurisdiction comply with legal requirements. (autorité réglementaire)
- site
site[Repealed, SOR/2002-368, s. 1]
- wholesale
wholesale[Repealed, SOR/2013-74, s. 2]
- wholesaler
wholesaler means a person who is not a distributor described in section C.01A.003 and who sells any of the following drugs other than at retail sale:
(a) a drug in dosage form that is listed in Schedule C or D to the Act, a drug that is a prescription drug or a controlled drug as defined in section G.01.001;
(b) an active ingredient;
(c) a narcotic as defined in the Narcotic Control Regulations; or
(d) a drug containing cannabis as defined in subsection 2(1) of the Cannabis Act. (grossiste)
(2) In this Division and in Division 2, drug does not include any of the following:
(a) a dilute drug premix;
(b) a medicated feed as defined in subsection 1(1) of the Feeds Regulations, 2024;
(c) an active ingredient that is for veterinary use and that is not an active pharmaceutical ingredient;
(d) an active pharmaceutical ingredient for veterinary use that is not required to be sold pursuant to a prescription and that is also a natural health product as defined in subsection 1(1) of the Natural Health Products Regulations;
(e) a drug that is used only for the purposes of an experimental study in accordance with a certificate issued under section C.08.015.
(3) Where the Minister designates additional dosage form classes, the Minister shall make a list of those classes available on request.
- SOR/97-12, s. 5
- SOR/98-7, s. 1
- SOR/2000-120, s. 1
- SOR/2002-368, s. 1
- SOR/2004-282, s. 1
- SOR/2013-74, s. 2
- SOR/2013-122, s. 14
- SOR/2017-76, s. 7
- SOR/2018-144, s. 367
- SOR/2019-171, s. 24
- SOR/2020-73, s. 1
- SOR/2024-132, s. 86
- SOR/2024-132, s. 87
Application
C.01A.002 (1) This Division does not apply to
(a) wholesaling a drug premix;
(b) subject to subsection (3), importing or compounding, pursuant to a prescription, a drug that is not commercially available in Canada by one of the following persons:
(i) a pharmacist,
(ii) a practitioner, and
(iii) a person who compounds a drug under the supervision of a practitioner;
(b.1) any activity with respect to a positron-emitting radiopharmaceutical that is used only for the purposes of a basic clinical research study described in section C.03.304;
(c) any activity with respect to a drug that is used only for the purposes of clinical testing in accordance with subsection C.05.006(1) or section C.08.005;
(d) fabricating, packaging/labelling, testing as required under Division 2, distributing as a distributer referred to in section C.01A.003, wholesaling or importing any of the following drugs for which prescriptions are not required and that are for human use in dosage form and not represented as a treatment, preventative or cure for any of the diseases, disorders or abnormal physical states set out in Schedule A.1 to the Act, namely,
(i) homeopathic drugs,
(ii) drugs that meet the requirements of a class monograph entitled “Vitamin Supplements”, “Mineral Supplements”, “Dietary Vitamin Supplements” or “Dietary Mineral Supplements”, as the case may be, and
(iii) drugs that
(A) contain a plant, mineral or animal substance in respect of which therapeutic activity or disease prevention activity is claimed, including traditional herbal medicines, traditional Chinese medicines, ayurvedic (East Indian) medicines and traditional aboriginal (North American) medicines, and
(B) the medical use of which is based solely on historical and ethnological evidence from references relating to a medical system other than one based on conventional scientific standards; and
(e) fabricating, packaging/labelling, testing, distributing, and importing of antimicrobial agents.
(1.1) This Division and Division 2 do not apply to a veterinary health product or an active pharmaceutical ingredient that is used in the fabrication of a veterinary health product.
(2) This Division and Divisions 2 to 4 do not apply to the affixing of a label to a previously labelled container.
(3) This Division applies to the importing, by a pharmacist, a veterinary practitioner or a person who compounds a drug under the supervision of a veterinary practitioner, of an active pharmaceutical ingredient for veterinary use that is for the purpose of compounding, pursuant to a prescription, a drug in dosage form that is not commercially available in Canada, if that ingredient is set out in List A.
- SOR/97-12, s. 5
- SOR/98-7, s. 2
- SOR/2001/-203, s. 1
- SOR/2004-282, s. 2
- SOR/2012-129, s. 1
- SOR/2017-76, s. 8
- SOR/2021-46, s. 10
C.01A.003 This Division and Divisions 2 to 4 apply to the following distributors:
(a) a distributor of an active ingredient; and
(b) a distributor of a drug for which the distributor holds the drug identification number.
- SOR/97-12, s. 5
- SOR/2002-368, s. 2
- SOR/2013-74, s. 3
- SOR/2017-259, s. 12
C.01A.003.1 For the purposes of this Division and the provisions of Divisions 2 to 4 that are prescribed in paragraphs A.01.048(b) to (d),
(a) a reference to a distributor referred to in section C.01A.003 or a distributor referred to in paragraph C.01A.003(a) includes a reference to a distributor of an active ingredient that is intended for use outside Canada; and
(b) a reference to a distributor referred to in section C.01A.003 or a distributor referred to in paragraph C.01A.003(b) includes a reference to a distributor of a drug in dosage form that is intended for consumption or use outside Canada.
Prohibition
C.01A.004 (1) Subject to subsection (2), no person shall, except in accordance with an establishment licence,
(a) fabricate, package/label or import a drug;
(b) perform the tests, including examinations, required under Division 2;
(c) distribute as a distributor referred to in section C.01A.003 a drug other than
(i) an active pharmaceutical ingredient, or
(ii) an active ingredient that is used in the fabrication of a drug that is of non-biological origin and that is listed in Schedule C to the Act; or
(d) wholesale a drug other than
(i) an active pharmaceutical ingredient, or
(ii) an active ingredient that is used in the fabrication of a drug that is of non-biological origin and that is listed in Schedule C to the Act.
(2) A person does not require an establishment licence to perform tests under Division 2 if the person holds an establishment licence as a fabricator, a packager/labeller, a distributor referred to in paragraph C.01A.003(b) or an importer.
(3) No person shall carry on an activity referred to in subsection (1) unless the person holds
(a) in respect of a narcotic as defined in the Narcotic Control Regulations, a licence for that narcotic under those Regulations;
(b) in respect of a controlled drug as defined in section G.01.001, a licence for that drug under Part G; or
(c) in respect of a drug containing cannabis as defined in subsection 2(1) of the Cannabis Act, a licence for that drug to conduct that activity under the Cannabis Regulations.
- SOR/97-12, s. 5
- SOR/2002-368, s. 3
- SOR/2013-74, s. 4
- SOR/2017-259, s. 13
- SOR/2018-144, s. 368
- SOR/2019-171, s. 24
- SOR/2022-100, s. 3
Application
- SOR/2011-81, s. 1(E)
C.01A.005 (1) A person who wishes to apply for an establishment licence shall submit an application to the Minister, in a form established by the Minister, that contains the following information and documents:
(a) the applicant’s name, address and telephone number, and their facsimile number and electronic mail address, if any;
(b) the name and telephone number, and the facsimile number and electronic mail address, if any, of a person to contact in case of an emergency;
(c) each activity set out in Table I to section C.01A.008 for which the licence is requested;
(d) each category of drugs set out in Table II to section C.01A.008 for which the licence is requested;
(e) each dosage form class in respect of which the applicant proposes to carry out a licensed activity, and whether it will be in a sterile dosage form;
(f) whether the applicant proposes to carry out a licensed activity in respect of an active ingredient;
(g) the address of each building in Canada in which the applicant proposes to fabricate, package/label, test as required under Division 2 or store drugs, specifying for each building the activities and the categories of drugs and, for each category, the dosage form classes, if any, and whether any drug will be in a sterile form;
(h) the address of each building in Canada at which records will be maintained;
(i) whether any building referred to in paragraphs (g) and (h) is a dwelling-house;
(j) the drug identification number, if any, or a name that clearly identifies the drug,
(i) for each narcotic as defined in the Narcotic Control Regulations, each controlled drug as defined in section G.01.001 or each drug containing cannabis as defined in subsection 2(1) of the Cannabis Act for which the licence is requested, and
(ii) for each other drug within a category of drugs for which the licence is requested, unless the licence is to perform tests required under Division 2, distribute as set out in paragraph C.01A.003(a), or wholesale;
(k) if any of the buildings referred to in paragraph (g) have been inspected under the Act or these Regulations, the date of the last inspection;
(l) evidence that the applicant’s buildings, equipment and proposed practices and procedures meet the applicable requirements of Divisions 2 to 4;
(m) in the case of an importer of a drug that is fabricated, packaged/labelled or tested in an MRA country at a recognized building,
(i) the name and address of each fabricator, packager/labeller and tester of the drug and the address of each building in which the drug is fabricated, packaged/labelled or tested, specifying for each building the activities and the categories of drugs and, for each category, the dosage form classes, if any, and whether any drug will be in a sterile form,
(ii) in respect of each activity done in an MRA country at a recognized building, the name of the regulatory authority that is designated under subsection C.01A.019(1) in respect of that activity for that drug and that has recognized that building as meeting its good manufacturing practices standards in respect of that activity for that drug, and
(iii) in respect of any other activities,
(A) a certificate from a Canadian inspector indicating that the fabricator’s, packager/labeller’s or tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4, or
(B) other evidence establishing that the fabricator’s, packager/labeller’s or tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4;
(n) in the case of any other importer, the name and address of each fabricator, packager/labeller and tester of the drugs proposed to be imported and the address of each building in which the drugs will be fabricated, packaged/labelled and tested, specifying for each building the activities and the categories of drugs and, for each category, the dosage form classes, if any, and whether any drug will be in a sterile form; and
(o) in the case of an importer referred to in paragraph (n),
(i) a certificate from a Canadian inspector indicating that the fabricator’s, packager/labeller’s and tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4, or
(ii) other evidence establishing that the fabricator’s, packager/labeller’s and tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4.
(2) In addition to the information and documents referred to in subsection (1), a person who submits an application for an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes a COVID-19 drug may include a statement to that effect in the application.
- SOR/97-12, s. 5
- SOR/2000-120, s. 2
- SOR/2002-368, s. 4
- SOR/2011-81, s. 2
- SOR/2013-74, s. 5
- SOR/2018-144, s. 369
- SOR/2019-171, s. 24
- SOR/2021-45, s. 3
C.01A.006 (1) A person who wishes to amend an establishment licence shall submit an application to the Minister, in a form established by the Minister, that contains the information and documents referred to in section C.01A.005 that relate to the amendment.
(1.1) In addition to the information and documents referred to in subsection (1), a person who submits an application to amend an establishment licence that relates to one or more activities set out in Table I to section C.01A.008 to be carried out in respect of a category of drugs set out in Table II to that section that includes a COVID-19 drug may include a statement to that effect in the application.
(2) An establishment licence must be amended where the licensee proposes
(a) to add an activity or category of drugs, as set out in the tables to section C.01A.008;
(b) in respect of a category of drugs and activity indicated in the licence, to authorize sterile dosage forms of the category;
(c) to add any building in Canada at which drugs are authorized to be fabricated, packaged/labelled, tested as required under Division 2 or stored, or to add, for an existing building, an authorization to fabricate, package/label, test or store a category of drugs, or sterile dosage forms of the category; and
(d) in addition to the matters set out in paragraphs (a) to (c), in the case of an importer,
(i) to add a fabricator, packager/labeller or tester of a drug,
(ii) to amend the name or address of a fabricator, packager/labeller or tester indicated in the licence, and
(iii) if the address of the buildings at which drugs are authorized to be fabricated, packaged/labelled or tested is indicated in the licence, to add additional buildings or, for an existing building, to add an authorization to fabricate, package/label or test a category of drugs, or sterile dosage forms of the category.
- SOR/97-12, s. 5
- SOR/2011-81, s. 3
- SOR/2021-45, s. 4
C.01A.007 (1) The Minister may, on receipt of an application for an establishment licence, an amendment to an establishment licence or the review of an establishment licence, require the applicant to submit further details pertaining to the information contained in the application that are necessary to enable the Minister to make a decision.
(2) When considering an application, the Minister may require that
(a) an inspection be made during normal business hours of any building referred to in paragraph C.01A.005(1)(g) or (h); and
(b) the applicant, if a fabricator, a packager/labeller, a person who performs tests required under Division 2, a distributor referred to in paragraph C.01A.003(b) or an importer, supply samples of any material to be used in the fabrication, packaging/labelling or testing of a drug.
- SOR/97-12, s. 5
- SOR/2011-81, s. 4
- SOR/2021-45, s. 5(F)
Issuance
C.01A.008 (1) Subject to subsection (1.1) and section C.01A.010, the Minister shall, on receipt of the information and material referred to in sections C.01A.005 to C.01A.007, issue or amend an establishment licence.
(1.1) The Minister shall, in determining whether he or she has received the information and material referred to in sections C.01A.005 to C.01A.007 in relation to an application referred to in subsection C.01A.005(2) or C.01A.006(1.1) that contains the statement referred to in the applicable subsection, also take into consideration the public health need related to COVID-19.
(2) The establishment licence shall indicate
(a) each activity set out in Table I to this section that is authorized and the category of drugs set out in Table II to this section for which each activity is authorized, specifying for each activity and category whether sterile dosage forms are authorized;
(b) the address of each building in Canada at which a category of drugs set out in Table II to this section is authorized to be fabricated, packaged/labelled, tested as required under Division 2 or stored, specifying for each building which of those activities and for which category of drugs, and whether sterile dosage forms of the category are authorized; and
(c) in addition to the matters referred to in paragraphs (a) and (b), in the case of an importer,
(i) the name and address of each fabricator, packager/labeller and tester from whom the importer is authorized to obtain the drug for import, and
(ii) the address of each building at which the drug is authorized to be fabricated, packaged/labelled or tested, specifying for each building the activities and the category of drugs set out in Table II to this section that are authorized, and whether sterile dosage forms are authorized.
(d) [Repealed, SOR/2002-368, s. 5]
(3) The Minister may indicate in an establishment licence a period for which records shall be retained under Division 2 that, based on the safety profile of the drug or materials, is sufficient to ensure the health of the consumer.
(4) When issuing an establishment licence, the Minister may impose terms and conditions on the establishment licence respecting
(a) the tests to be performed in respect of a drug, and the equipment to be used, to ensure that the drug is not unsafe for use; and
(b) any other matters necessary to prevent risk to the health of consumers, including conditions under which drugs are fabricated, packaged/labelled or tested.
TABLE I
Item Activities 1 Fabricate 2 Package/label 3 Perform the tests, including any examinations, required under Division 2 4 Distribute as a distributor referred to in paragraph C.01A.003(a) an active ingredient other than (a) an active pharmaceutical ingredient; or
(b) an active ingredient that is used in the fabrication of a drug that is of non-biological origin and that is listed in Schedule C to the Act
5 Distribute as a distributor referred to in paragraph C.01A.003(b) 6 Import 7 Wholesale a drug other than (a) an active pharmaceutical ingredient; or
(b) an active ingredient that is used in the fabrication of a drug that is of non-biological origin and that is listed in Schedule C to the Act
TABLE II
Item Categories of drugs 1 Pharmaceuticals 1.1 Active ingredients 2 Vaccines 3 [Repealed, SOR/2013-179, s. 2] 4 Drugs that are listed in Schedule D to the Act, other than vaccines 5 Drugs listed in Schedule C to the Act 6 Drugs that are prescription drugs, controlled drugs as defined in section G.01.001, narcotics as defined in the Narcotic Control Regulations and drugs containing cannabis as defined in subsection 2(1) of the Cannabis Act
7 Active pharmaceutical ingredients set out in List A that are for veterinary use
- SOR/97-12, s. 5
- SOR/2000-120, s. 3
- SOR/2002-368, s. 5
- SOR/2013-74, s. 6
- SOR/2013-122, s. 15
- SOR/2013-179, s. 2
- SOR/2017-76, s. 9
- SOR/2017-259, s. 14
- SOR/2018-144, s. 370
- SOR/2019-171, s. 24
- SOR/2021-45, s. 6
- SOR/2021-46, s. 11(E)
- SOR/2022-100, s. 4
Annual Licence Review
C.01A.009 (1) The holder of an establishment licence that is not suspended shall submit an application for the review of their licence to the Minister before April 1 of each year and include with it the information and documents referrred to in section C.01A.005.
(2) The Minister shall conduct an annual review of the licence on the basis of the information and documents submitted by the holder and any other relevant information in the Minister’s possession.
- SOR/97-12, s. 5
- SOR/97-298, s. 1
- SOR/2011-81, s. 5
Refusal to Issue
C.01A.010 (1) The Minister may refuse to issue or amend an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) if
(a) the applicant has made a false or misleading statement in relation to the application for the licence; or
(b) the applicant has had an establishment licence suspended in respect of the matter.
(2) The Minister shall refuse to issue or amend an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) if the Minister has reasonable grounds to believe that issuing or amending an establishment licence in respect of the matter would constitute a risk to the health of the consumer.
(3) Where the Minister refuses to issue or amend an establishment licence, the Minister shall
(a) notify the applicant in writing of the reasons for the refusal; and
(b) give the applicant an opportunity to be heard.
- SOR/97-12, s. 5
Terms and Conditions
C.01A.011 (1) Every person who holds an establishment licence shall comply with
(a) the requirements of the establishment licence; and
(b) the applicable requirements of Divisions 2 to 4.
(2) [Repealed, SOR/2000-120, s. 4]
- SOR/97-12, s. 5
- SOR/2000-120, s. 4
- SOR/2021-45, s. 7
C.01A.012 (1) The Minister may amend the terms and conditions of an establishment licence that are imposed under subsection C.01A.008(4) if the Minister believes on reasonable grounds that an amendment is necessary to prevent risk to the health of the consumer.
(2) The Minister shall give at least 15 days notice in writing to the holder of the establishment licence of the proposed amendment, the reasons for it and its effective date.
- SOR/97-12, s. 5
- SOR/2021-45, s. 8
- SOR/2021-46, s. 11(E)
C.01A.012.1 (1) Despite subsection C.01A.008(4), the Minister may, at any time, including when issuing an establishment licence, impose terms and conditions on an establishment licence that is issued or amended under section C.01A.008 on the basis of an application referred to in subsection C.01A.005(2) or C.01A.006(1.1) that contains the statement referred to in the applicable subsection.
(2) For greater certainty, terms and conditions that may be imposed under subsection (1) are not limited to those that may be imposed under subsection C.01A.008(4).
C.01A.012.2 The Minister may, at any time, amend terms or conditions that are imposed on an establishment licence under subsection C.01A.012.1(1).
Notification
C.01A.013 Every person who holds an establishment licence shall notify the Minister in writing within 15 days after
(a) there is any change to the information referred to in any of paragraphs C.01A.005(1)(a), (b) and (e) to (i); or
(b) an event occurs that results in their being in contravention of any of the applicable requirements of Divisions 2 to 4, where it may affect the quality, safety or efficacy of a drug fabricated, packaged/labelled, tested as required under Division 2 or stored by them.
- SOR/97-12, s. 5
- SOR/2017-18, s. 15
- SOR/2021-45, s. 10
C.01A.014 (1) No licensee shall carry on a licensed activity in respect of any category of drugs if a change referred to in subsection (2) has occurred in respect of that category, unless
(a) they have filed with the Minister a notice that contains sufficient information to enable the Minister to assess the safety of the drug, taking into account the change; and
(b) the Minister has issued to them a letter indicating that the information will be reviewed and has not, within 90 days after issuing the letter, sent them a notice indicating that the change is not acceptable.
(2) Notification is required in respect of the following changes where they may affect whether a drug can be fabricated, packaged/labelled, tested or stored in accordance with the applicable requirements of Divisions 2 to 4:
(a) changes to the plans and specifications of a building where a drug is fabricated, packaged/labelled, tested or stored;
(b) changes to the equipment that is used in the fabrication, packaging/labelling or testing of a drug;
(c) changes to the practices or procedures; and
(d) in the case of an importer, other than an importer of a drug that is fabricated, packaged/labelled or tested in an MRA country at a recognized building, any change referred to in paragraphs (a) to (c) that relates to the fabricator, packager/labeller or tester of the drug being imported.
- SOR/97-12, s. 5
- SOR/2000-120, s. 5
- SOR/2002-368, s. 6
C.01A.015 (1) An importer of a drug that is fabricated, packaged/labelled or tested in an MRA country at a recognized building shall immediately notify the Minister if the fabricator, packager/labeller or tester indicated in the importer’s establishment licence no longer holds a valid permit, licence or other authorization issued by the regulatory authority that recognized that building.
(2) The Minister shall, on receiving a notification under subsection (1), amend the importer’s establishment licence by removing the name and address of that fabricator, packager/labeller or tester.
- SOR/97-12, s. 5
- SOR/2000-120, s. 6
- SOR/2002-368, s. 7
Suspension
C.01A.016 (1) The Minister may suspend an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) if he or she has reasonable grounds to believe that
(a) the licensee has contravened any provision of the Act or these Regulations; or
(b) the licensee has made a false or misleading statement in the application for the establishment licence.
(2) Before suspending an establishment licence, the Minister shall consider
(a) the licensee’s history of compliance with the Act and these Regulations; and
(b) the risk that allowing the licence to continue in force would constitute for the health of the consumer.
(3) The Minister shall not suspend an establishment licence until
(a) the Minister has sent the licensee a written notice that sets out the reason for the proposed suspension, any corrective action required to be taken and the time within which it must be taken;
(b) if corrective action is required, the time set out in the notice has passed without the action having been taken; and
(c) the licensee has been given an opportunity to be heard in respect of the suspension.
- SOR/97-12, s. 5
- SOR/2018-84, s. 4
- SOR/2021-46, s. 7
C.01A.017 (1) The Minister may suspend an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) without giving the licensee an opportunity to be heard if it is necessary to do so to prevent a risk to the health of consumers, by giving the licensee a notice that states the reason for the suspension.
(2) A licensee may request of the Minister, in writing, that the suspension be reconsidered.
(3) The Minister shall, within 45 days after the date of receiving the request, provide the licensee with the opportunity to be heard.
- SOR/97-12, s. 5
- SOR/2018-84, s. 5
C.01A.017.1 The Minister may suspend an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) if, after the Minister has, under section 21.31 of the Act, ordered the licensee to conduct an assessment in order to provide evidence establishing that the licensee’s buildings, equipment or practices and procedures, as the case may be, continue to meet the requirements referred to in paragraph C.01A.005(1)(l), subparagraph C.01A.005(1)(m)(ii) or (iii) or paragraph C.01A.005(1)(o),
(a) the licensee fails to comply with the order; or
(b) the licensee complies with the order but the Minister determines that the results of the assessment are not sufficient to establish that those requirements continue to be met.
- SOR/2018-84, s. 6
- SOR/2021-45, s. 11
C.01A.018 The Minister shall reinstate an establishment licence in respect of any or all matters indicated in subsection C.01A.008(2) that are the subject of the suspension if, within 12 months after the effective date of the suspension, the licensee provides the Minister with sufficient evidence demonstrating that
(a) the situation on which the suspension was based has been corrected; or
(b) the situation on which the suspension was based did not exist.
- SOR/97-12, s. 5
- SOR/2018-84, s. 6
Cancellation
C.01A.018.1 The Minister shall cancel an establishment licence if the licensee has failed to submit an application for the review of the licence in accordance with subsection C.01A.009(1).
- SOR/2011-81, s. 6
- SOR/2018-84, s. 7
C.01A.018.2 (1) If the Minister has suspended an establishment licence in respect of all matters indicated in subsection C.01A.008(2) and the suspension is still in effect 12 months after the effective date of the suspension, the Minister shall cancel the licence.
(2) If the Minister has suspended an establishment licence in respect of one or more of the matters indicated in subsection C.01A.008(2) and the suspension is still in effect 12 months after the effective date of the suspension, the Minister shall cancel the licence only in respect of the matters that are the subject of the suspension.
- SOR/2018-84, s. 7
Designation
C.01A.019 (1) For the purposes of this Division and Divisions 2 to 4, a regulatory authority that is set out in column 1 of the List of Regulatory Authorities for the Purposes of Section C.01A.019 of the Food and Drug Regulations, published by the Government of Canada on its website, as amended from time to time, is designated in respect of the activities set out in column 3 for the drugs or category of drugs set out in column 2.
(2) Whole blood and its components are excluded from the drugs and categories of drugs that are set out in column 2 of the List.
(3) The lot release of drugs listed in Schedule D to the Act is excluded from the activity of testing set out in column 3 of the List.
- SOR/97-12, s. 5
- SOR/2000-120, s. 7
- SOR/2002-368, s. 8
- SOR/2024-136, s. 2
DIVISION 2Good Manufacturing Practices
C.02.001 [Repealed, SOR/97-12, s. 5.1]
C.02.002 In this Division,
- drug
drug[Repealed, SOR/97-12, s. 6]
- importer
importer[Repealed, SOR/97-12, s. 6]
- medical gas
medical gas means any gas or mixture of gases manufactured, sold or represented for use as a drug; (gaz médical)
- packaging material
packaging material includes a label; (matériel d’emballage)
- produce
produce[Repealed, SOR/97-12, s. 6]
- quality control department
quality control department[Repealed, SOR/2010-95, s. 1]
- specifications
specifications means a detailed description of a drug, the raw material used in a drug or the packaging material for a drug and includes
(a) a statement of all properties and qualities of the drug, raw material or packaging material that are relevant to the manufacture, packaging and use of the drug, including the identity, potency and purity of the drug, raw material or packaging material,
(b) a detailed description of the methods used for testing and examining the drug, raw material or packaging material, and
(c) a statement of tolerances for the properties and qualities of the drug, raw material or packaging material. (spécifications)
- SOR/82-524, s. 3
- SOR/85-754, s. 1
- SOR/89-174, s. 1
- SOR/97-12, s. 6
- SOR/2010-95, s. 1
C.02.002.1 This Division does not apply to fabricating, packaging/labelling, testing, storing and importing of antimicrobial agents.
- SOR/2004-282, s. 3
C.02.002.2 In this Division,
(a) a reference to specifications is — in respect of a drug intended for consumption or use outside Canada, the raw material used in such a drug or the packaging material for such a drug — a reference to the specifications with which the drug, raw material or packaging material is required to comply in the country in which the drug is intended to be consumed or used; and
(b) the definition expiration date in subsection C.01.001(1) does not apply in respect of a drug intended for consumption or use outside Canada.
Sale
C.02.003 No distributor referred to in paragraph C.01A.003(b) and no importer shall sell a drug unless it has been fabricated, packaged/labelled, tested and stored in accordance with the requirements of this Division.
- SOR/82-524, s. 3
- SOR/97-12, s. 7
- SOR/2000-120, s. 8
- SOR/2010-95, s. 2(F)
C.02.003.1 No person shall sell a drug that they have fabricated, packaged/labelled, tested or stored unless they have fabricated, packaged/labelled, tested or stored it in accordance with the requirements of this Division.
- SOR/2013-74, s. 7
C.02.003.2 (1) No person shall import an active ingredient into Canada for the purpose of sale unless they have in Canada a person who is responsible for its sale.
(2) No person who imports an active ingredient into Canada shall sell any lot or batch of it unless the following appear on its label:
(a) the name and civic address of the person who imports it; and
(b) the name and address of the principal place of business in Canada of the person who is responsible for its sale.
- SOR/2013-74, s. 7
Use in Fabrication
C.02.003.3 No person shall use an active ingredient in the fabrication of a drug unless it is fabricated, packaged/labelled, tested and stored in accordance with the requirements of this Division.
- SOR/2013-74, s. 7
Premises
C.02.004 The premises in which a lot or batch of a drug is fabricated, packaged/labelled or stored shall be designed, constructed and maintained in a manner that
(a) permits the operations therein to be performed under clean, sanitary and orderly conditions;
(b) permits the effective cleaning of all surfaces therein; and
(c) prevents the contamination of the drug and the addition of extraneous material to the drug.
- SOR/82-524, s. 3
- SOR/97-12, s. 8
- SOR/2010-95, s. 3
Equipment
C.02.005 The equipment with which a lot or batch of a drug is fabricated, packaged/labelled or tested shall be designed, constructed, maintained, operated and arranged in a manner that
(a) permits the effective cleaning of its surfaces;
(b) prevents the contamination of the drug and the addition of extraneous material to the drug; and
(c) permits it to function in accordance with its intended use.
- SOR/82-524, s. 3
- SOR/97-12, s. 9
Personnel
C.02.006 Every lot or batch of a drug shall be fabricated, packaged/labelled, tested and stored under the supervision of personnel who, having regard to the duties and responsibilities involved, have had such technical, academic and other training as the Minister considers satisfactory in the interests of the health of the consumer or purchaser.
- SOR/82-524, s. 3
- SOR/85-754, s. 2
- SOR/97-12, s. 52
- SOR/2018-69, s. 27
Sanitation
C.02.007 (1) Every person who fabricates or packages/labels a drug shall have a written sanitation program that shall be implemented under the supervision of qualified personnel.
(2) The sanitation program referred to in subsection (1) shall include
(a) cleaning procedures for the premises where the drug is fabricated or packaged/labelled and for the equipment used in the fabrication or packaging/labelling; and
(b) instructions on the sanitary fabrication and packaging/labelling of drugs and the handling of materials used in the fabrication and packaging/labelling of drugs.
- SOR/82-524, s. 3
- SOR/97-12, ss. 10, 53
C.02.008 (1) Every person who fabricates or packages/labels a drug shall have, in writing, minimum requirements for the health and the hygienic behaviour and clothing of personnel to ensure the clean and sanitary fabrication and packaging/labelling of the drug.
(2) No person shall have access to any area where a drug is exposed during its fabrication or packaging/labelling if the person
(a) is affected with or is a carrier of a disease in a communicable form; or
(b) has an open lesion on any exposed surface of the body.
- SOR/82-524, s. 3
- SOR/97-12, s. 11
Raw Material Testing
C.02.009 (1) Each lot or batch of raw material shall be tested against the specifications for that raw material prior to its use in the fabrication of a drug.
(2) No lot or batch of raw material shall be used in the fabrication of a drug unless that lot or batch of raw material complies with the specifications for that raw material.
(3) Notwithstanding subsection (1), water may, prior to the completion of its tests under that subsection, be used in the fabrication of a drug.
(4) Where any property of a raw material is subject to change on storage, no lot or batch of that raw material shall be used in the fabrication of a drug after its storage unless the raw material is retested after an appropriate interval and complies with its specifications for that property.
(5) Where the specifications referred to in subsections (1), (2) and (4) are not prescribed, they shall
(a) be in writing;
(b) be acceptable to the Minister who shall take into account the specifications contained in any publication mentioned in Schedule B to the Act; and
(c) be approved by the person in charge of the quality control department.
- SOR/82-524, s. 3
- SOR/97-12, s. 59
- SOR/2018-69, s. 27
C.02.010 (1) The testing referred to in section C.02.009 shall be performed on a sample taken
(a) after receipt of each lot or batch of raw material on the premises of the fabricator; or
(b) subject to subsection (2), before receipt of each lot or batch of raw material on the premises of the fabricator, if
(i) the fabricator
(A) has evidence satisfactory to the Minister to demonstrate that raw materials sold to him by the vendor of that lot or batch of raw material are consistently manufactured in accordance with and consistently comply with the specifications for those raw materials, and
(B) undertakes periodic complete confirmatory testing with a frequency satisfactory to the Minister, and
(ii) the raw material has not been transported or stored under conditions that may affect its compliance with the specifications for that raw material.
(2) After a lot or batch of raw material is received on the premises of the fabricator, the lot or batch of raw material shall be tested for identity.
- SOR/82-524, s. 3
- SOR/97-12, ss. 12, 60
- SOR/2018-69, s. 27
Manufacturing Control
C.02.011 (1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) and importer of a drug shall have written procedures prepared by qualified personnel in respect of the drug to ensure that the drug meets the specifications for that drug.
(2) Every person required to have written procedures referred to in subsection (1) shall ensure that each lot or batch of the drug is fabricated, packaged/labelled and tested in compliance with those procedures.
- SOR/82-524, s. 3
- SOR/97-12, s. 13
C.02.012 (1) Every fabricator, packager/labeller, distributor referred to in section C.01A.003, importer and wholesaler of a drug shall maintain
(a) a system of control that permits complete and rapid recall of any lot or batch of the drug that is on the market; and
(b) a program of self-inspection.
(2) Every fabricator and packager/labeller and, subject to subsections (3) and (4), every distributor referred to in paragraph C.01A.003(b) and importer of a drug shall maintain a system to ensure that any lot or batch of the drug fabricated and packaged/labelled on premises other than their own is fabricated and packaged/labelled in accordance with the requirements of this Division.
(3) Subsection (2) does not apply to a distributor if the drug is fabricated, packaged/labelled and tested in Canada by a person who holds an establishment licence that authorizes those activities in respect of that drug.
(4) Subsection (2) does not apply to a distributor or importer if the drug is fabricated or packaged/labelled in an MRA country at a recognized building and both of the following requirements are met:
(a) the address of the building is set out in their establishment licence; and
(b) they retain a copy of the batch certificate for each lot or batch of the drug that they receive.
- SOR/82-524, s. 3
- SOR/97-12, s. 13
- SOR/2000-120, s. 9
- SOR/2002-368, s. 9
- SOR/2013-74, s. 8
Quality Control Department
C.02.013 (1) Every fabricator, packager/labeller, wholesaler, distributor referred to in section C.01A.003 and importer of a drug shall have on their premises in Canada a quality control department that is supervised by personnel described in section C.02.006.
(2) Except in the case of a wholesaler or a distributor referred to in paragraph C.01A.003(a), the quality control department shall be a distinct organizational unit that functions and reports to management independently of any other functional unit, including the manufacturing, processing, packaging or sales unit.
- SOR/82-524, s. 3
- SOR/89-174, s. 8(F)
- SOR/97-12, s. 55
- SOR/2000-120, s. 10
- SOR/2010-95, s. 4
- SOR/2013-74, s. 9
C.02.014 (1) Except in the case of a wholesaler or a distributor referred to in paragraph C.01A.003(a), no lot or batch of a drug shall be made available for further use in fabrication or for sale unless the person in charge of the quality control department approves the sale or the further use.
(2) A drug that is returned to its fabricator, packager/labeller, wholesaler, distributor referred to in section C.01A.003 or importer shall not be made available for further use in fabrication or for further sale unless the person in charge of the quality control department approves the further sale or further use.
(3) No lot or batch of a raw material or packaging/labelling material shall be used in the fabrication or packaging/labelling of a drug unless the person in charge of the quality control department approves the use.
(4) No lot or batch of a drug shall be reprocessed unless the person in charge of the quality control department approves the reprocessing.
- SOR/82-524, s. 3
- SOR/89-174, s. 8(F)
- SOR/97-12, ss. 14, 55
- SOR/2010-95, s. 5
- SOR/2013-74, s. 9
C.02.015 (1) All fabrication, packaging/labelling, testing, storage and transportation methods and procedures that may affect the quality of a drug shall be examined and approved by the person in charge of the quality control department before their implementation.
(2) The person in charge of the quality control department shall cause to be investigated any complaint or information that is received respecting the quality of a drug or its deficiencies or hazards and cause any necessary corrective action to be taken, in the case where the complaint or information relates to an activity over which the department exercises quality control.
(2.1) In the case where the complaint or information that is received does not relate to an activity over which the quality control department exercises quality control, the person in charge of the department shall forward the complaint or information to the person in charge of the quality control department that exercises quality control over that activity.
(3) The person in charge of the quality control department shall cause all tests or examinations required pursuant to this Division to be performed by a competent laboratory.
- SOR/82-524, s. 3
- SOR/97-12, s. 15
- SOR/2010-95, s. 6
Packaging Material Testing
C.02.016 (1) Each lot or batch of packaging material shall, prior to its use in the packaging of a drug, be examined or tested against the specifications for that packaging material.
(2) No lot or batch of packaging material shall be used in the packaging of a drug unless the lot or batch of packaging material complies with the specifications for that packaging material.
(3) The specifications referred to in subsections (1) and (2) shall
(a) be in writing;
(b) be acceptable to the Minister who shall take into account the specifications contained in any publication mentioned in Schedule B to the Act; and
(c) be approved by the person in charge of the quality control department.
- SOR/82-524, s. 3
- SOR/89-174, s. 8(F)
- SOR/2018-69, s. 27
C.02.017 (1) The examination or testing referred to in section C.02.016 shall be performed on a sample taken
(a) after receipt of each lot or batch of packaging material on the premises of the person who packages a drug; or
(b) subject to subsection (2), before receipt of each lot or batch of packaging material on the premises of the person who packages a drug, if
(i) that person
(A) has evidence satisfactory to the Minister to demonstrate that packaging materials sold to him by the vendor of that lot or batch of packaging material are consistently manufactured in accordance with and consistently comply with the specifications for those packaging materials, and
(B) undertakes periodic complete confirmatory examination or testing with a frequency satisfactory to the Minister,
(ii) the packaging material has not been transported or stored under conditions that may affect its compliance with the specifications for that packaging material.
(2) After a lot or batch of packaging material is received on the premises of the person who packages a drug,
(a) the lot or batch of the packaging material shall be examined or tested for identity; and
(b) the labels shall be examined or tested in order to ensure that they comply with the specifications for those labels.
- SOR/82-524, s. 3
- SOR/89-174, ss. 2(F), 8(F)
- SOR/97-12, s. 56(F)
- SOR/2018-69, s. 27
Finished Product Testing
C.02.018 (1) Each lot or batch of a drug shall, before it is made available for further use in fabrication or for sale, be tested against the specifications for that drug.
(2) No lot or batch of a drug shall be made available for further use in fabrication or for sale unless it complies with the specifications for that drug.
(3) The specifications referred to in subsections (1) and (2) shall
(a) be in writing;
(b) be approved by the person in charge of the quality control department; and
(c) comply with the Act and these Regulations.
- SOR/82-524, s. 3
- SOR/2013-74, s. 10
C.02.019 (1) A packager/labeller of a drug, a distributor referred to in paragraph C.01A.003(b) and an importer of a drug other than an active ingredient shall perform the finished product testing on a sample of the drug that is taken either
(a) after receipt of each lot or batch of the drug on their premises in Canada; or
(b) before receipt of each lot or batch of the drug on their premises in Canada if the following conditions are met:
(i) the packager/labeller, distributor or importer
(A) has evidence satisfactory to the Minister to demonstrate that drugs sold to them by the vendor of that lot or batch are consistently manufactured in accordance with and consistently comply with the specifications for those drugs, and
(B) undertakes periodic complete confirmatory testing, with a frequency satisfactory to the Minister, and
(ii) the drug has not been transported or stored under conditions that may affect its compliance with the specifications for that drug.
(2) If the packager/labeller, distributor or importer receives a lot or batch of a drug on their premises in Canada the useful life of which is more than 30 days, the lot or batch shall be tested for identity and the packager/labeller shall confirm the identity after the lot or batch is packaged/labelled.
(3) Subsections (1) and (2) do not apply to a distributor if the drug is fabricated, packaged/labelled and tested in Canada by a person who holds an establishment licence that authorizes that activity.
(4) Subsections (1) and (2) do not apply to a distributor or importer if the drug is fabricated, packaged/labelled and tested in an MRA country at a recognized building and they retain a copy of the batch certificate for each lot or batch of the drug that they receive.
(4.1) Subsections (1) and (2) do not apply to a distributor or importer of a COVID-19 drug if it is the subject of a written request made under section C.04.015.
(4.2) Subsection (1) does not apply to a packager/labeller, distributor or importer if the following conditions are met:
(a) the drug is a radiopharmaceutical or a radionuclide generator, as defined in section C.03.001, and has a useful life of no more than 30 days; and
(b) the packager/labeller, distributor or importer maintains evidence satisfactory to the Minister to demonstrate that
(i) the drug has been tested against the specifications for that drug, and
(ii) the drug has not been transported or stored under conditions that may affect its compliance with those specifications.
(4.3) Subsections (1) and (2) do not apply to a packager/labeller, distributor or importer if the following conditions are met:
(a) the drug is
(i) for human use,
(ii) in dosage form,
(iii) listed in Schedule D to the Act,
(iv) not a vaccine, and
(v) composed of genetically modified autologous human nucleated cells or is delivered to cells using an adeno-associated virus vector; and
(b) the packager/labeller, distributor or importer maintains evidence satisfactory to the Minister to demonstrate that
(i) the drug has been tested against the specifications for that drug, and
(ii) the drug has not been transported or stored under conditions that may affect its compliance with those specifications.
(5) Subsections (1) and (2) do not apply to a distributor or importer of a drug that is not a prescription drug and that is part of a class of drugs that is set out in column 1 of the List of Non-prescription Drugs Not Subject to Certain Testing Requirements if all of the following conditions are met:
(a) the drug contains, as its only active ingredients, one or more of those set out in column 2, each of which corresponds to that class, in the corresponding quantity set out in column 3, and the drug is consistent with the descriptive information set out in columns 4 to 6;
(b) the drug is
(i) fabricated in Canada or in a recognized country or region,
(ii) packaged/labelled in Canada or in a recognized country or region, and
(iii) tested in a recognized country or region;
(c) the distributor or the importer retains a copy of the batch certificate for each lot or batch of the drug that is distributed or imported, as the case may be.
(6) A drug that is referred to in subsection (4.2), (4.3) or (5) and that is imported may be shipped directly to a person other than the importer if the following conditions are met:
(a) before importing the drug, the importer receives a document that demonstrates that the drug complies with the specifications for that drug;
(b) the importer and the distributor have measures in place to ensure that all the requirements of these Regulations in respect of the importation of the drug are met.
- SOR/82-524, s. 3
- SOR/89-174, s. 8(F)
- SOR/97-12, ss. 16, 57
- SOR/2000-120, s. 11
- SOR/2002-368, s. 10
- SOR/2013-74, s. 11
- SOR/2018-69, s. 27
- SOR/2020-73, s. 2
- SOR/2021-45, s. 12
- SOR/2024-136, s. 4
Records
C.02.020 (1) Every fabricator, packager/labeller, distributor referred to in paragraph C.01A.003(b) and importer shall maintain all of the following records on their premises in Canada for each drug that they fabricate, package/label, distribute or import:
(a) except in the case of an importer of an active pharmaceutical ingredient or an active ingredient that is used in the fabrication of a drug that is of non-biological origin and that is listed in Schedule C to the Act, master production documents for the drug;
(b) evidence that each lot or batch of the drug has been fabricated, packaged/labelled, tested and stored in accordance with the procedures described in the master production documents;
(c) evidence that the conditions under which the drug was fabricated, packaged/labelled, tested and stored are in compliance with the requirements of this Division;
(d) evidence that establishes the period during which the drug in the container in which it is sold or made available for further use in fabrication will meet the specifications for that drug; and
(e) evidence that the finished product testing referred to in section C.02.018 was carried out and the results of that testing.
(2) Every distributor referred to in paragraph C.01A.003(b) and importer shall make available to the Minister, on request, the results of testing performed on raw materials and packaging/labelling materials for each lot or batch of a drug that it distributes or imports.
(3) Every fabricator shall maintain on their premises written specifications for all raw materials and adequate evidence of the testing of those raw materials referred to in section C.02.009 and of the test results.
(4) Every person who packages a drug shall maintain on their premises written specifications for all packaging materials and adequate evidence of the examination or testing of those materials referred to in section C.02.016 and of any test results.
(5) Every fabricator, packager/labeller and tester shall maintain on their premises in Canada detailed plans and specifications of each building in Canada where they fabricate, package/label or test drugs and a description of the design and construction of those buildings.
(6) Every fabricator, packager/labeller and tester shall maintain on their premises in Canada personnel records in respect of each person who is employed to supervise the fabrication, packaging/labelling and testing of drugs, including the person’s title, responsibilities, qualifications, experience and training.
- SOR/82-524, s. 3
- SOR/89-174, ss. 3(F), 8(F)
- SOR/97-12, ss. 17, 52, 60
- SOR/2013-74, s. 11
- SOR/2017-259, s. 15
- SOR/2018-69, s. 27
C.02.021 (1) All records and evidence of the fabrication, packaging/labelling, finished product testing referred to in section C.02.018 and storage of a drug in dosage form that are required to be maintained under this Division shall be retained for one year after the expiration date of the drug unless the person’s establishment licence specifies some other period.
(2) Subject to subsection (4), all records and evidence of the fabrication, packaging/labelling, finished product testing referred to in section C.02.018 and storage of an active ingredient that are required to be maintained under this Division shall be retained in respect of each lot or batch of the active ingredient for the following period unless the person holds an establishment licence that specifies some other period:
(a) in the case of an active ingredient that has a retest date, three years after the lot or batch has been completely distributed; or
(b) in any other case, one year after the expiration date of the lot or batch.
(3) Subject to subsection (4), all records and evidence of the raw material testing referred to in section C.02.009 and of the testing of packaging/labelling materials that are required to be maintained under this Division shall be retained for five years after the raw materials and packaging/labelling materials were last used in the fabrication or packaging/labelling of a drug unless the person’s establishment licence specifies some other period.
(4) If a fabricator is required to maintain records and evidence in respect of the same active ingredient under subsections (2) and (3), they shall maintain them for the longest period that is applicable.
- SOR/82-524, s. 3
- SOR/89-174, s. 8(F)
- SOR/92-654, s. 6
- SOR/97-12, s. 18
- SOR/2013-74, s. 11
C.02.022 (1) Every wholesaler, distributor referred to in section C.01A.003 and importer of a drug in dosage form shall retain records of sale of each lot or batch of the drug, which enable them to recall the lot or batch from the market, for one year after the expiration date of that lot or batch unless their establishment licence specifies some other period.
(2) Every distributor of an active ingredient referred to in paragraph C.01A.003(a) and every wholesaler and importer of an active ingredient shall retain records of sale of each lot or batch of the active ingredient, which enable them to recall the lot or batch from the market, for the following period unless the person holds an establishment licence that specifies some other period:
(a) in the case of an active ingredient that has a retest date, three years after the lot or batch has been completely distributed; or
(b) in any other case, one year after the expiration date of the lot or batch.
- SOR/82-524, s. 3
- SOR/92-654, s. 7
- SOR/97-12, s. 18
- SOR/2013-74, s. 11
C.02.023 (1) On receipt of a complaint or any information respecting the quality of a drug or its deficiencies or hazards, every fabricator, packager/labeller, wholesaler, distributor referred to in section C.01A.003 and importer of the drug shall make a record of the complaint or information that contains the following:
(a) the results of any investigation carried out under subsection C.02.015(2) and, if applicable, the corrective action taken; or
(b) the name and business address of the person in charge of the quality control department to whom the complaint or information was forwarded under subsection C.02.015(2.1) and the date on which it was forwarded.
(2) Records referred to in subsection (1) shall be retained for the following period unless the person holds an establishment licence that specifies some other period:
(a) in the case of a drug in dosage form, one year after the expiration date of the lot or batch of the drug; and
(b) in the case of an active ingredient,
(i) if the active ingredient has a retest date, three years after the lot or batch has been completely distributed, or
(ii) in any other case, one year after the expiration date of the lot or batch of the active ingredient.
- SOR/82-524, s. 3
- SOR/92-654, s. 7
- SOR/97-12, s. 18
- SOR/2010-95, s. 7
- SOR/2013-74, s. 11
C.02.024 (1) Every fabricator, packager/labeller, distributor referred to in section C.01A.003, importer and wholesaler shall
(a) maintain records of the results of the self-inspection program required by section C.02.012 and of any action taken in connection with that program; and
(b) retain those records for a period of at least three years.
(2) Every person who fabricates or packages/labels a drug shall
(a) maintain records on the operation of the sanitation program required to be implemented under section C.02.007; and
(b) retain those records for a period of at least three years.
- SOR/82-524, s. 3
- SOR/97-12, ss. 19, 53
C.02.024.1 Every distributor of an active ingredient referred to in paragraph C.01A.003(a) and every fabricator, packager/labeller, wholesaler and importer of an active ingredient shall add all of the following information to the documentation that accompanies the active ingredient, immediately after any like information that has been added by another person:
(a) their establishment licence number, or if there is none, their name, address, telephone number, fax number and email address;
(b) an indication whether they have fabricated, packaged/labelled, wholesaled, distributed or imported the active ingredient and the date on which that activity was carried out;
(c) the expiration date; and
(d) the lot number.
- SOR/2013-74, s. 12
Samples
C.02.025 (1) Every distributor referred to in paragraph C.01A.003(b) and importer of a drug in dosage form shall retain in Canada a sample of each lot or batch of the packaged/labelled drug for one year after the expiration date of the drug unless their establishment licence specifies otherwise.
(2) Subject to subsection (4), the fabricator of a drug in dosage form shall retain a sample of each lot or batch of raw materials used in the fabrication for two years after the materials were last used in the fabrication unless their establishment licence specifies some other period.
(3) Subject to subsection (4), the fabricator of an active ingredient shall retain a sample of each lot or batch of it for the following period unless their establishment licence specifies some other period:
(a) in the case of an active ingredient that has a retest date, three years after the lot or batch has been completely distributed; or
(b) in any other case, one year after the expiration date of the lot or batch.
(4) If a fabricator is required to maintain samples in respect of the same active ingredient under subsections (2) and (3), they shall maintain them for the longest period that is applicable.
- SOR/82-524, s. 3
- SOR/89-174, s. 4(F)
- SOR/92-654, s. 8
- SOR/97-12, s. 20
- SOR/2013-74, s. 13
- SOR/2021-46, s. 8
C.02.026 The samples referred to in section C.02.025 shall be in an amount that is sufficient to determine whether the drug or raw material complies with the specifications for that drug or raw material.
- SOR/82-524, s. 3
Stability
C.02.027 (1) Every distributor referred to in paragraph C.01A.003(b) and importer of a drug in dosage form shall establish the period during which each drug in the package in which it is sold will comply with the specifications for that drug.
(2) Every fabricator and importer of an active ingredient shall establish the period during which each drug in the package in which it is sold will comply with the specifications for that drug.
- SOR/82-524, s. 3
- SOR/97-12, s. 58
- SOR/2013-74, s. 14
C.02.028 (1) Every distributor referred to in paragraph C.01A.003(b) and importer of a drug in dosage form shall monitor, by means of a continuing program, the stability of the drug in the package in which it is sold.
(2) Every fabricator and importer of an active ingredient shall monitor, by means of a continuing program, the stability of the drug in the package in which it is sold.
- SOR/82-524, s. 3
- SOR/97-12, s. 58
- SOR/2013-74, s. 14
Sterile Products
C.02.029 In addition to the other requirements of this Division, a drug that is intended to be sterile shall be fabricated and packaged/labelled
(a) in separate and enclosed areas;
(b) under the supervision of personnel trained in microbiology; and
(c) by a method scientifically proven to ensure sterility.
- SOR/82-524, s. 3
- SOR/97-12, s. 21
Medical Gases
C.02.030 The provisions of sections C.02.025, C.02.027 and C.02.028 do not apply to medical gases.
- SOR/85-754, s. 3
DIVISION 3
Schedule C Drugs
C.03.001 In this Division,
- drug
drug means a drug that is listed in Schedule C to the Act that is in dosage form or a drug that is an active ingredient of biological origin that can be used in the preparation of a drug listed in that Schedule; (drogue)
- licence
licence or Canadian licence[Repealed, SOR/97-12, s. 22]
- manufacturer
manufacturer[Repealed, SOR/97-12, s. 22]
- master lot
master lot means a quantity of a drug from which a lot is prepared for sale by subsequent dilution or mixture; (maître-lot)
- radionuclide generator
radionuclide generator means a radioactive parent and daughter
(a) contained in an ion-exchange column, or
(b) dissolved in a suitable solvent in a liquid-liquid extraction system
where the radioactive daughter is separated from its parent by
(c) elution from the ion exchange column, or
(d) a solvent extraction procedure. (générateur de radionucléide)
- SOR/97-12, s. 22
- SOR/2013-74, s. 15
C.03.001.1 No distributor referred to in paragraph C.01A.003(b) or importer shall sell a drug unless it has been fabricated, packaged/labelled, tested and stored in accordance with this Division.
- SOR/97-12, s. 23
C.03.002 to C.03.005 [Repealed, SOR/97-12, s. 24]
C.03.006 [Repealed, SOR/97-12, s. 67]
C.03.007 to C.03.011 [Repealed, SOR/97-12, s. 26]
C.03.012 On written request from the Minister, every fabricator, packager/labeller, tester, distributor referred to in paragraph C.01A.003(b) and importer of a drug shall submit protocols of tests together with samples of any lot or master lot of the drug before it is sold, and no person shall sell a lot of which the protocol or sample fails to meet the requirements of these Regulations.
- SOR/97-12, s. 27
- SOR/2018-69, s. 27
C.03.013 No person shall fabricate or import a drug that is derived from animal tissue unless the tissue is obtained from a healthy animal free from infectious disease.
- SOR/97-12, s. 27
C.03.014 (1) Section C.01.004 does not apply to a drug.
(2) and (3) [Repealed, SOR/97-12, s. 28]
- SOR/79-236, s. 1
- SOR/93-202, s. 15
- SOR/97-12, s. 28
C.03.015 (1) Every package of a drug that is a prescription drug shall carry the symbol “Pr” on the upper left quarter of the principal display panel of both its inner and outer labels or, in the case of a single dose container, on the upper left quarter of its outer label.
(2) Subsection (1) does not apply to
(a) a drug sold to a drug fabricator;
(b) a drug sold under a prescription;
(c) a radiopharmaceutical as defined in section C.03.201; or
(d) a component or kit as defined in section C.03.205.
- SOR/80-543, s. 9
- SOR/97-12, s. 61
- SOR/2001-181, s. 4
- SOR/2013-122, s. 16
C.03.030 to C.03.045 [Repealed, SOR/81-335, s. 2]
Radiopharmaceuticals
C.03.201 In these Regulations, radiopharmaceutical means a drug that exhibits spontaneous disintegration of unstable nuclei with the emission of nuclear particles or photons.
- SOR/97-12, s. 29
C.03.202 (1) Every package containing a radiopharmaceutical, other than a radionuclide generator, shall carry,
(a) on both the inner and the outer labels,
(i) the proper name of the drug, which proper name, where there is a brand name, shall immediately precede or follow the brand name,
(ii) the name of the distributor referred to in paragraph C.01A.003(b),
(iii) the lot number, and
(iv) the drug identification number assigned for the radiopharmaceutical, preceded by the expression “Drug Identification Number” or “Drogue : identification numérique”, or both, or the abbreviation “DIN”; and
(b) on the outer label
(i) the address of the distributor referred to in paragraph C.01A.003(b),
(ii) the standard that the drug professes to meet, if that standard is referred to in any publication mentioned in Schedule B to the Act,
(iii) a statement of the pharmaceutical form or the route of administration of the drug,
(iv) a statement of the recommended use and the recommended radioactivity to be administered for that use, or a reference to an accompanying package insert that shows such information,
(v) [Repealed, SOR/2017-259, s. 16]
(vi) the radiation warning symbol set out in Schedule 3 to the Radiation Protection Regulations and the words “RAYONNEMENT — DANGER — RADIATION”,
(vii) the names and a statement of the amounts of any preservatives or stabilizing agents contained in the drug,
(viii) the names and a statement of the amounts of all other non-radioactive contents of the drug,
(ix) a statement of the total radioactivity content of the drug including overfill,
(x) a statement of the total volume of the drug including overfill, except where its contents are entirely in gaseous, capsule or lyophilized form,
(xi) a statement of the concentration of radioactive material in the drug expressed as
(A) units of radioactivity per capsule or
(B) units of radioactivity per unit volume,
except where the contents of the drug are entirely in gaseous or lyophilized form,
(xii) a statement of the specific activity of the drug expressed as units of radioactivity per unit weight of carrier present or the statement “carrier-free” or “sans entraîneur”, whichever is applicable,
(xiii) a statement of the reference time in respect of the radioactivity values mentioned in subparagraphs (ix), (xi) and (xii), the name of the month being written or designated by letter abbreviation,
(xiv) a statement of the recommended useful life or the date after which the drug is not recommended for use, the name of the month being written or designated by letter abbreviation, and
(xv) a statement of the special storage requirements with reference to temperature and light.
(2) Subparagraph (1)(a)(iv) does not apply to a radiopharmaceutical that is
(a) compounded by a pharmacist under a prescription or by a practitioner; or
(b) sold under a prescription, if the radiopharmaceutical’s label indicates
(i) its proper name, common name or brand name,
(ii) its potency, and
(iii) the name of its manufacturer.
(3) Subparagraph (1)(b)(viii) of this section does not apply where the information referred to in that subparagraph is shown on a package insert that accompanies the drug.
(4) Section C.01.005 does not apply to a radiopharmaceutical.
- SOR/79-236, s. 2
- SOR/93-202, s. 16
- SOR/97-12, ss. 54, 58, 62
- SOR/2001-203, s. 2
- SOR/2012-129, s. 2
- SOR/2017-259, s. 16
- SOR/2018-69, s. 35(F)
C.03.203 (1) Every radionuclide generator shall carry on the inner label
(a) the proper name of the radionuclide generator, which proper name, where there is a brand name, shall immediately precede or follow the brand name;
(b) the name and address of the distributor referred to in paragraph C.01A.003(b);
(c) the lot number;
(d) the standard that the radionuclide generator professes to meet, if that standard is referred to in any publication mentioned in Schedule B to the Act;
(e) the drug identification number assigned for the radionuclide generator, preceded by the expression “Drug Identification Number” or “Drogue : identification numérique”, or both, or the abbreviation “DIN”;
(f) the radiation warning symbol set out in Schedule 3 to the Radiation Protection Regulations and the words “RAYONNEMENT — DANGER — RADIATION”;
(g) a statement of the total parent radioactivity contained in the radionuclide generator;
(h) a statement of the hour and date at which the radioactivity value mentioned in paragraph (g) is valid, the name of the month being written or designated by letter abbreviation;
(i) a statement of the recommended useful life or the date after which the radionuclide generator is not recommended for use, the name of the month being written or designated by letter abbreviation;
(j) a statement of the recommended useful life of the drug after removal from the radionuclide generator;
(k) a statement of special storage requirements with reference to temperature or shielding;
(l) complete directions for use or a reference to an accompanying package insert that sets out such directions; and
(m) a statement cautioning against the dismantling of the radionuclide generator.
(2) Paragraphs (1)(i) and (j) of this section do not apply where the information referred to in those subparagraphs is shown on a package insert that accompanies the radionuclide generator.
(3) Section C.01.005 does not apply to a radionuclide generator.
- SOR/79-236, s. 3
- SOR/93-202, s. 17
- SOR/97-12, ss. 54, 58, 62
- SOR/2012-129, s. 3
- SOR/2017-259, s. 17
- SOR/2018-69, ss. 21(F), 22(F)
C.03.204 (1) No person shall sell a drug that contains technetium-99m at any time during its useful life if it also contains a radionuclidic impurity set out in the monograph for Sodium Pertechnetate Tc-99m Injection referred to in the publication set out in item 8 of Schedule B to the Act, in an amount greater than that shown in the monograph.
(2) No person shall sell a radionuclide generator from which can be removed a drug that contains technetium-99m, at any time during the useful life of the drug, if the drug also contains a radionuclidic impurity set out in the monograph for Sodium Pertechnetate Tc-99m Injection referred to in the publication set out in item 8 of Schedule B to the Act, in an amount greater than that shown in the monograph.
- SOR/97-12, s. 30
- SOR/2012-129, s. 4
Drugs, Other than Radionuclides, Sold or Represented for Use in the Preparation of Radiopharmaceuticals
- SOR/2017-259, s. 18(F)
C.03.205 The following definitions apply in this section and in sections C.03.206 to C.03.209.
- component
component means
(a) a unit of a drug, other than a radionuclide, separately packaged in a kit; or
(b) an empty vial or other accessory item in a kit. (constituant)
- kit
kit means a package that is intended to be used in the preparation of radiopharmaceuticals and that
(a) contains one or more separately packaged units of a drug, other than a radionuclide; and
(b) may contain empty vials or other accessory items. (trousse)
- SOR/79-236, s. 4
- SOR/2017-259, s. 19
C.03.206 Sections C.01.005 and C.04.019 do not apply to a component or kit.
- SOR/79-236, s. 4
C.03.207 (1) Every component shall be labelled to show
(a) adequate identification of the component and an adequate description of its function;
(b) where applicable, a quantitative list of its ingredients or a reference to the label of the kit that shows such information;
(c) the name of the distributor referred to in paragraph C.01A.003(b);
(d) the lot number;
(e) a statement of any special storage requirements with respect to temperature and light;
(f) the date after which the component is not recommended for use, the name of the month being written in full or designated by letter abbreviation; and
(g) adequate directions for use or a reference to the accompanying package insert that shows such directions.
(2) The component of a kit that is intended to contain the prepared radiopharmaceutical shall be labelled to display the drug identification number assigned for the kit, preceded by the expression “Drug Identification Number” or “Drogue : identification numérique”, or both, or the abbreviation “DIN”.
- SOR/79-236, s. 4
- SOR/97-12, s. 58
- SOR/2017-259, s. 20
- SOR/2018-69, s. 23(F)
C.03.208 Every kit shall be labelled to show
(a) its proper name;
(b) its brand name, if any;
(c) a list of its contents;
(d) the name and address of the distributor referred to in paragraph C.01A.003(b);
(e) the drug identification number assigned for the kit, preceded by the expression “Drug Identification Number” or “Drogue : identification numérique”, or both, or the abbreviation “DIN”;
(f) the lot number;
(g) a statement of any special storage requirements with respect to temperature and light;
(h) the date after which the kit is not recommended for use, the name of the month being written in full or designated by letter abbreviation;
(i) where the label of a component makes reference to the label of the kit that shows information as to the ingredients of the component, a quantitative list of the ingredients of that component;
(j) a statement of the sterility and apyrogenicity of the components;
(k) adequate directions for preparing the radiopharmaceutical or a reference to the accompanying package insert that shows such directions;
(l) a statement of the duration of the useful life of the prepared radiopharmaceutical;
(m) a statement of the storage requirements for the prepared radiopharmaceutical;
(n) a statement of the recommended use for the prepared radiopharmaceutical and the recommended radioactivity to be administered for that use, or a reference to the accompanying package insert that shows such information; and
(o) a statement of the route of administration of the prepared radiopharmaceutical.
(p) [Repealed, SOR/2001-203, s. 3]
- SOR/79-236, s. 4
- SOR/93-202, s. 18
- SOR/97-12, ss. 58, 62
- SOR/2001-203, s. 3
- SOR/2017-259, s. 21
- SOR/2018-69, s. 36(F)
C.03.209 A package insert shall be included in every kit and shall show
(a) the proper name and the brand name, if any, of the kit and a description of its use;
(b) a list of the contents of the kit;
(c) the name and address of the distributor referred to in paragraph C.01A.003(b) of the kit;
(d) identification of the radionuclides that can be used to prepare the radiopharmaceutical;
(e) directions for preparing the radiopharmaceutical and a statement of the storage requirements for the prepared radiopharmaceutical;
(f) a statement of the duration of the useful life of the prepared radiopharmaceutical;
(g) a description of the biological actions of the prepared radiopharmaceutical;
(h) indications and contraindications in respect of the prepared radiopharmaceutical;
(i) warnings and precautions in respect of the components and the prepared radiopharmaceutical;
(j) the adverse reactions, if any, associated with the prepared radiopharmaceutical;
(k) where applicable, the pharmacology and toxicology of the prepared radiopharmaceutical or a statement that such information is available on request;
(l) the radiation dosimetry in respect of the prepared radiopharmaceutical;
(m) a statement of the recommended use for the prepared radiopharmaceutical and the recommended radioactivity to be administered for that use;
(n) a statement of the route of administration of the prepared radiopharmaceutical; and
(o) a recommendation that the radiochemical purity and radioactivity content of the prepared radiopharmaceutical be checked prior to administration.
- SOR/79-236, s. 4
- SOR/93-202, s. 19
- SOR/97-12, s. 58
- SOR/2018-69, s. 36(F)
Positron-emitting Radiopharmaceuticals
Interpretation
C.03.301 The following definitions apply in this section and in sections C.03.302 to C.03.319.
- adverse reaction
adverse reaction means an undesirable and unintended response in a study subject or other person to a study drug that is caused by the administration of any dose of the study drug. (réaction indésirable)
- good clinical practices
good clinical practices means generally accepted clinical practices that are designed to protect the rights, safety and well-being of study subjects and other persons. (bonnes pratiques cliniques)
- import
import means, in respect of a study drug, to import it into Canada for sale for the purpose of a study. (importer)
- other person
other person means an individual who comes into physical contact with a study subject. (autre personne)
- protocol
protocol means a document that describes the objectives, design, methodology, statistical considerations and organization of a study. (protocole)
- qualified investigator
qualified investigator means the physician and member in good standing of a professional medical association in Canada to whom a sponsor gives the responsibility for the proper conduct of the study at a given study site, who is entitled to practise their profession under the laws of the province where the study site is located. (chercheur qualifié)
- research ethics board
research ethics board means a body described in section C.03.306. (comité d’éthique de la recherche)
- serious adverse reaction
serious adverse reaction means an adverse reaction that results in any of the following consequences for the study subject or other person:
(a) their in-patient hospitalization or its prolongation;
(b) a congenital malformation;
(c) persistent or significant disability or incapacity;
(d) a life-threatening condition; or
(e) death. (réaction indésirable grave)
- serious unexpected adverse reaction
serious unexpected adverse reaction means a serious adverse reaction that is not identified in nature, severity or frequency in the risk information set out on the label of the study drug. (réaction indésirable grave et imprévue)
- sponsor
sponsor means a person who is responsible for the conduct of a study. (promoteur)
- study
study means a basic clinical research study that involves human subjects and that is described in sections C.03.304 and C.03.305. (étude)
- study drug
study drug means a positron-emitting radiopharmaceutical that is used in a study. (drogue destinée à l’étude)
- study site
study site means the location where all or part of a study is conducted. (lieu d’étude)
- SOR/2012-129, s. 5
Application
C.03.302 (1) Sections C.03.303 to C.03.319 apply to the sale and importation of study drugs.
(2) Sections C.03.001 to C.03.209 and Divisions 5 and 8 do not apply to study drugs.
(3) Sections C.03.303 to C.03.319 do not apply to a study drug manufactured from a bulk process intermediate that is of biological origin.
- SOR/2012-129, s. 5
Prohibition
C.03.303 No person shall sell or import a study drug unless all of the following requirements are met:
(a) the study drug is for use only in a study;
(b) the study drug has been previously tested in human subjects and its safety in humans has been demonstrated;
(c) if the study drug is to be imported, the manufacturer of the drug has a representative in Canada who is responsible for its sale;
(d) the sponsor is authorized under section C.03.309 to sell or import the study drug; and
(e) the sponsor complies with sections C.03.310 to C.03.316.
- SOR/2012-129, s. 5
Purpose of Study
C.03.304 (1) The purpose of a study is to obtain data on any of the following:
(a) the pharmacokinetics or metabolism of the study drug;
(b) normal human biochemistry or physiology; or
(c) changes caused to human biochemistry or physiology by aging, disease or medical interventions.
(2) A study is not primarily intended to do any of the following:
(a) discover, identify or verify the pharmacodynamic effects of the study drug;
(b) identify adverse reactions;
(c) fulfil an immediate therapeutic or diagnostic purpose; or
(d) ascertain the safety or efficacy of the study drug.
- SOR/2012-129, s. 5
Requirements
C.03.305 (1) A study shall meet all of the following requirements:
(a) before the study drug is used in the study, there is sufficient data from testing it in animals and humans to demonstrate its safety in humans;
(b) the amount of active ingredients or combination of active ingredients in the study drug has been shown not to cause any clinically detectable pharmacodynamic effect in humans;
(c) the total radiation dose incurred annually by a study subject, including from multiple administrations of the study drug, from significant contaminants or from impurities and from the use of other procedures for the purposes of the study, will be not more than 50 mSv;
(d) any concomitant drug used in the study has been assigned a drug identification number under subsection C.01.014.2(1) or, in the case of a concomitant drug that is a new drug, has been issued a notice of compliance under section C.08.004;
(e) study subjects shall be at least 18 years old and have legal capacity at the time of the study;
(f) female study subjects shall
(i) be confirmed at the outset of the study, on the basis of a pregnancy test, as not being pregnant or state in writing that they are not pregnant, and
(ii) be advised that if they are lactating, they are to suspend lactation for 24 hours after the administration of the study drug; and
(g) the study shall not involve more than 30 study subjects.
(2) Despite paragraph (1)(g), a study may involve more than 30 study subjects if the sponsor provides the Minister with a scientific rationale for the increase and the Minister approves it.
- SOR/2012-129, s. 5
Research Ethics Board
C.03.306 A research ethics board has all of the following characteristics:
(a) its principal mandate is to approve the initiation of and to periodically review biomedical research that involves human subjects in order to protect their rights, safety and well-being;
(b) it has at least five members, a majority of whom are Canadian citizens or permanent residents under the Immigration and Refugee Protection Act, is composed of both men and women and includes at least the following:
(i) two members whose primary experience and expertise are in a scientific discipline, who have broad experience in the methods and areas of research to be approved and one of whom is from a medical discipline,
(ii) one member knowledgeable in ethics,
(iii) one member knowledgeable in Canadian laws relevant to the research to be approved,
(iv) one member whose primary experience and expertise are in a non-scientific discipline, and
(v) one member who is from the community or is a representative of an organization interested in the areas of research to be approved and who is not affiliated with the sponsor or with the study site; and
(c) it has no affiliations with the sponsor that could compromise its ability to fulfil its principal mandate, or that could be perceived to do so.
- SOR/2012-129, s. 5
Application for Authorization
C.03.307 (1) The sponsor shall submit to the Minister an application for authorization to sell or import a study drug that contains the information set out in subsection (2) as well as sufficient information to demonstrate that all of the following criteria are met:
(a) the use of the study drug will not endanger the health of any study subject or other person;
(b) the study is not contrary to the best interests of the study subjects; and
(c) the objectives of the study can reasonably be achieved.
(2) The application shall contain all of the following information:
(a) the title of the study and the protocol code or identification;
(b) the purposes and a concise description of the study;
(c) the number of study subjects;
(d) the brand name, if any, of the study drug;
(e) the chemical or generic name of the active ingredients in the study drug;
(f) a qualitative list of the non-active ingredients of the study drug;
(g) the maximum mass of the study drug to be administered to each study subject;
(h) the radioactive dose range of the study drug, expressed in MBq or mCi;
(i) the effective dose or effective dose equivalent of the study drug, expressed in mSv/MBq or rem/mCi;
(j) the sponsor’s name and civic address, its postal address if different, and its telephone number, fax number and email address;
(k) the manufacturer’s name and civic address, its postal address if different, and its telephone number, fax number and email address;
(l) in the case of an application for importation, the name and civic address, the postal address if different, and the telephone number, fax number and email address of the manufacturer’s representative in Canada who is responsible for the sale of the study drug;
(m) the name and civic address of each study site;
(n) for each study site, the name, civic address, telephone number, fax number and email address of the qualified investigator;
(o) the proposed starting date for the study at each study site, if known;
(p) for each study site, the name, civic address, telephone number, fax number and email address of the research ethics board;
(q) a statement, dated and signed by the research ethics board for each study site, that certifies that it has reviewed and approved the study, the protocol and the statement of the risks and anticipated benefits arising to the health of study subjects as a result of participating in the study that is set out in the informed consent form;
(r) a list of any previous applications for an authorization to sell or import a drug for a study related to the current study; and
(s) a statement, dated and signed by the sponsor’s senior medical or scientific officer in Canada and senior executive officer, that certifies both of the following:
(i) the study will be conducted in accordance with these Regulations, and
(ii) all of the information contained or referred to in the application is complete and accurate and is not false or misleading.
- SOR/2012-129, s. 5
Additional Information
C.03.308 If the information submitted under section C.03.307 is insufficient to enable the Minister to determine whether the sale or importation of the study drug should be authorized, the Minister may, by notice in writing, request the sponsor to provide any additional information that is necessary to make the determination and that is relevant to the study drug, the study or the protocol, by the date specified in the notice.
- SOR/2012-129, s. 5
Authorization
C.03.309 After examining the application and any additional information, the Minister shall authorize the sponsor to sell or import the study drug if she or he determines that the application complies with the requirements of section C.03.307, and shall send a notice of that decision to the sponsor that specifies the study sites in respect of which the sale or importation are authorized.
- SOR/2012-129, s. 5
Notice
C.03.310 The sponsor shall notify the Minister in writing of the day on which the sale or importation of the study drug is intended to start in respect of each study site, not later than 15 days before that day.
- SOR/2012-129, s. 5
Good Clinical Practices
C.03.311 A sponsor shall ensure that each study is conducted in accordance with good clinical practices and that
(a) the study is scientifically sound and clearly described in its protocol;
(b) the study is conducted, and the study drug is used, in accordance with the protocol and with these Regulations;
(c) systems and procedures are implemented that assure the quality of every aspect of the study;
(d) at each study site, there is only one qualified investigator;
(e) at each study site, medical care and medical decisions, in respect of the study, are under the supervision of the qualified investigator;
(f) each individual who is involved in the conduct of the study is qualified by their education, training and experience to perform their respective tasks;
(g) before a study subject participates in the study, a copy of their signed consent form is included in the records for the study;
(h) the requirements respecting information and records set out in section C.03.315 are met; and
(i) the study drug is manufactured, handled and stored in accordance with Division 2, other than sections C.02.019, C.02.025 and C.02.026.
- SOR/2012-129, s. 5
Labelling
C.03.312 Despite any other provision of these Regulations respecting labelling, the sponsor shall ensure that the study drug
(a) bears an inner label that sets out both of the following:
(i) the unique batch number for the study drug, and
(ii) the radiation warning symbol set out in Schedule 3 to the Radiation Protection Regulations and the words “RAYONNEMENT — DANGER — RADIATION”; and
(b) is accompanied by a package insert that sets out all of the following information:
(i) a statement that indicates that the study drug is to be used only under the supervision of a qualified investigator,
(ii) the chemical or generic name of the active ingredients in the study drug,
(iii) the name and civic address of the manufacturer,
(iv) the name and civic address of the sponsor,
(v) the code or other identification of the protocol,
(vi) the warnings and precautions in respect of the use of the study drug, and
(vii) a list of the possible adverse reactions that are associated with the use of the study drug.
- SOR/2012-129, s. 5
- SOR/2018-69, s. 24(F)
- SOR/2021-46, s. 9(F)
Submission of Information
C.03.313 (1) On the Minister’s written request, a sponsor shall submit, within the period specified in the request, information to establish the safety of the study drug if the Minister has reason to believe any of the following:
(a) the use of the study drug may endanger the health of a study subject or other person;
(b) the study may be contrary to the best interests of the study subjects;
(c) a qualified investigator is not respecting their undertaking made under paragraph C.03.315(3)(f); or
(d) information submitted in respect of the study drug or study is false or misleading.
(2) The Minister may, by notice in writing, require the sponsor to provide the Minister with any information or records referred to in subsection C.03.315(3) to assess the safety of the study drug or the health of the study subjects or other persons, by the date specified in the notice.
- SOR/2012-129, s. 5
Adverse Reaction Reporting
C.03.314 (1) During the course of a study, the sponsor shall notify the Minister of any serious adverse reaction or serious unexpected adverse reaction that occurs inside or outside Canada, within the following period:
(a) if the adverse reaction is fatal or life-threatening, within seven days after becoming aware of it; or
(b) if the adverse reaction is not fatal or life-threatening, within 15 days after becoming aware of it.
(2) The sponsor shall, within eight days after having notified the Minister under subsection (1), file with the Minister a complete report in respect of the adverse reaction, including an assessment of the importance and implication of the findings.
(3) Sections C.01.016 to C.01.020 do not apply to study drugs.
- SOR/2012-129, s. 5
Records
C.03.315 (1) The sponsor shall record, handle and store all information in respect of a study in a way that allows it to be reported completely and accurately and to be interpreted and verified.
(2) The sponsor shall maintain complete and accurate records to establish that the study is conducted in accordance with these Regulations.
(3) The sponsor shall maintain all of the following records in respect of the use of the study drug in each study:
(a) records respecting all adverse reactions that occur inside or outside Canada, including the indications for use and the dosage form of the study drug at the time of the adverse reaction;
(b) written procedures for subject monitoring and for the documentation and reporting of adverse reactions;
(c) articles from scientific journals or other publications that were used in support of the safety profile of the study drug in respect of humans;
(d) records in respect of each study subject, including respecting their enrolment, a copy of their signed consent form and sufficient information to enable them to be identified and contacted in the event that the sale of the study drug may endanger their health or that of another person;
(e) records respecting the shipment, receipt, sale, return and destruction or other disposition of the study drug;
(f) for each study site, an undertaking, dated and signed by the qualified investigator before the start of the study, that they will
(i) conduct the study in accordance with good clinical practices, and
(ii) on discontinuance of the study by the sponsor, for any reason related to health or safety, immediately inform both the study subjects and the research ethics board of the discontinuance, provide them with the reasons for the discontinuance and advise them in writing of any potential risks to the health of study subjects or other persons;
(g) for each study site, a copy of the informed consent form; and
(h) for each study site, a copy of the certifying statement described in paragraph C.03.307(2)(q), of the protocol for the study and of the statement of the risks and anticipated benefits arising to the health of study subjects as a result of participating in the study that is set out in the informed consent form.
(4) The sponsor shall maintain all records for five years after the day on which the study ends.
- SOR/2012-129, s. 5
Discontinuance of a Study
C.03.316 (1) If a sponsor discontinues a study in its entirety or at a study site, the sponsor shall notify all qualified investigators of the discontinuance as soon as possible in writing, and include in the notice the reasons for the discontinuance and whether the study presented any risks to the health of study subjects or other persons.
(2) If the discontinuance is for reasons that would affect the health or safety of study subjects or other persons, the sponsor shall notify the Minister in writing within 15 days after the discontinuance, and include in the notice the reasons for the discontinuance and whether it will have an impact on any proposed or ongoing studies in respect of the study drug in Canada by the sponsor.
- SOR/2012-129, s. 5
Suspension
C.03.317 (1) The Minister shall suspend an authorization to sell or import a study drug, in its entirety or in respect of a study site, in any of the following circumstances:
(a) information provided by the sponsor under section C.03.307, C.03.308 or C.03.313 proves to be inaccurate or incomplete;
(b) the sponsor fails to provide the Minister with sufficient information to establish the safety of the study drug pursuant to a written request under section C.03.313, by the date specified in the request;
(c) the sponsor fails to notify the Minister of an adverse reaction or file a report in respect of an adverse reaction in accordance with section C.03.314; or
(d) the sponsor contravenes a provision of these Regulations or any provision of the Act in relation to the study drug.
(2) In determining whether to suspend an authorization in its entirety or in respect of a study site, the Minister shall consider whether the reason for the suspension affects the study in its entirety or affects only a certain study site.
(3) Before suspending an authorization, the Minister shall send the sponsor a notice that
(a) specifies whether the suspension is of the study authorization in its entirety or in respect of a study site and sets out the reasons for the proposed suspension and the effective date;
(b) if applicable, specifies the corrective action that the sponsor must take and the period within which it must be taken; and
(c) gives the sponsor a reasonable opportunity to be heard in writing concerning the proposed suspension.
(4) Despite subsection (3), the Minister shall immediately suspend an authorization if she or he has reason to believe that it is necessary to do so to prevent injury to the health of a study subject or any other person.
(5) When the Minister suspends an authorization under subsection (4), the Minister must send the sponsor a notice that
(a) sets out the reasons for the suspension;
(b) if applicable, specifies the corrective action that the sponsor must take and the period within which it must be taken; and
(c) gives the sponsor a reasonable opportunity to be heard in writing concerning the suspension.
- SOR/2012-129, s. 5
Reinstatement
C.03.318 (1) Subject to subsection (2), the Minister shall reinstate the authorization if the sponsor provides the Minister with sufficient evidence to establish that the study does not present a risk of injury to the health of study subjects or other persons, within the following periods:
(a) in the case of a suspension under subsection C.03.317(1), 30 days after the day on which the suspension is effective; or
(b) in the case of a suspension under subsection C.03.317(4), the period specified in the notice sent under subsection C.03.317(5).
(2) If the Minister does not reinstate any part of an authorization that was suspended, the Minister shall amend the authorization to remove that part.
- SOR/2012-129, s. 5
Cancellation
C.03.319 (1) The Minister shall cancel an authorization, in its entirety or in respect of a study site, in either of the following circumstances:
(a) the study is discontinued in its entirety or at that study site by the sponsor under section C.03.316; or
(b) the sponsor fails to provide the Minister with the evidence required by subsection C.03.318(1) within the specified period.
(2) When the Minister cancels all or part of an authorization, she or he shall send the sponsor a notice that sets out the reasons for the cancellation and the effective date.
- SOR/2012-129, s. 5
DIVISION 4
Schedule D Drugs
C.04.001 In this Division,
- date of manufacture
date of manufacture means
(a) in the case of a product for which a standard of potency exists, the date it satisfactorily passes a potency test,
(b) in the case of an animal product for which no standard of potency exists, the date of its removal from the animal, and
(c) in the case of a product other than an animal product for which no standard of potency exists, the date of cessation of growth; (date de fabrication)
- drug
drug means a drug that is listed in Schedule D to the Act that is in dosage form or a drug that is an active ingredient that can be used in the preparation of a drug listed in that Schedule; (drogue)
- licence
licence or Canadian licence[Repealed, SOR/97-12, s. 31]
- manufacturer
manufacturer [Repealed, SOR/97-12, s. 31]
- SOR/97-12, s. 31
- SOR/2013-74, s. 16
C.04.001.1 No distributor referred to in paragraph C.01A.003(b) or importer shall sell a drug unless it has been fabricated, packaged/labelled, tested and stored in accordance with this Division.
- SOR/97-12, s. 32
C.04.002 This Division does not apply to a drug in oral dosage form that contains micro-organisms if the drug is recommended solely for restoring, normalizing or stabilizing the intestinal flora.
- SOR/97-12, s. 33
C.04.003 The date of issue of a drug shall be the date on which the finished product is removed from cold storage but in any case shall be, not later than
(a) six months after the date of manufacture for a drug that has been kept constantly at a temperature not exceeding 10°C;
(b) 12 months after the date of manufacture for a drug that has been kept constantly at a temperature not exceeding 5°C; or
(c) two years after the date of manufacture for a drug that has been kept constantly at a temperature not exceeding 0°C.
C.04.004 to C.04.006 [Repealed, SOR/97-12, s. 34]
C.04.007 [Repealed, SOR/97-12, s. 67]
C.04.008 to C.04.012 [Repealed, SOR/97-12, s. 36]
C.04.013 Every fabricator and packager/labeller shall safely segregate all work with spore-bearing, pathogenic micro-organisms and other infectious agents known to require special precautions in manipulation and shall take such care of equipment and arrangements for supervision that the possibility of contamination of other drugs is avoided.
- SOR/97-12, s. 63
C.04.014 No person shall conduct laboratory procedures of a diagnostic nature in their premises unless those procedures are entirely segregated from the fabrication, packaging/labelling and testing of drugs.
- SOR/97-12, s. 37
C.04.015 On written request from the Minister, every fabricator, packager/labeller, tester, distributor referred to in paragraph C.01A.003(b) and importer of a drug shall submit protocols of tests together with samples of any lot of the drug before it is sold, and no person shall sell any lot of that drug if the protocol or sample fails to meet the requirements of these Regulations.
- SOR/97-12, s. 37
- SOR/2018-69, s. 27
C.04.016 All animals from which drugs are prepared and preserved shall be
(a) under the direct supervision of competent medical or veterinary personnel;
(b) kept in quarantine by the fabricator for at least seven days before use; and
(c) healthy and free from infectious disease.
- SOR/97-12, s. 38
C.04.017 A fabricator shall keep necropsy records of all animals that die or are killed after having been used in the production of a drug.
- SOR/97-12, s. 61
C.04.018 A fabricator shall immediately segregate, and report the fact to the Minister, any animal with actual or suspected vesicular stomatitis, foot and mouth disease, encephalomyelitis, infectious anaemia, glanders, anthrax, tetanus or any other serious infectious disease.
- SOR/97-12, s. 61
C.04.019 The provisions of section C.01.004 do not apply to a drug as defined in this Division but every package of such drug shall carry
(a) on both the inner and the outer labels
(i) the proper name of the drug, which proper name, where there is a brand name, shall immediately precede or follow the brand name in type not less than one-half the size of that of the brand name,
(ii) the name of the distributor referred to in paragraph C.01A.003(b),
(iii) the potency of the drug, where applicable,
(iv) the recommended dose of the drug,
(v) the lot number,
(vi) the expiration date except upon the inner label of a single-dose container, and
(vii) adequate direction for use; and
(b) on the outer label
(i) the address of the distributor referred to in paragraph C.01A.003(b),
(ii) [Repealed, SOR/2013-179, s. 3]
(iii) the proper name, or the common name if there is no proper name, and the amount, of any preservative in the drug,
(iv) a statement that the drug shall be stored at a temperature of not less than 2°C and not more than 10°C, unless the Minister has received evidence demonstrating that such a statement is not required,
(v) a statement of the net contents in terms of weight, measure, or number, and
(vi) in the case of a new drug for extraordinary use in respect of which a notice of compliance has been issued under section C.08.004.01, the following statement, displayed in capital letters and in a legible manner:
“HEALTH CANADA HAS AUTHORIZED THE SALE OF THIS EXTRAORDINARY USE NEW DRUG FOR [naming purpose] BASED ON LIMITED CLINICAL TESTING IN HUMANS.
SANTÉ CANADA A AUTORISÉ LA VENTE DE CETTE DROGUE NOUVELLE POUR USAGE EXCEPTIONNEL AUX FINS DE [indication de la fin] EN SE FONDANT SUR DES ESSAIS CLINIQUES RESTREINTS CHEZ L’ÊTRE HUMAIN.”.
- SOR/78-424, s. 7
- SOR/93-202, s. 21
- SOR/97-12, ss. 39, 54, 58
- SOR/2011-88, s. 6
- SOR/2013-179, s. 3
C.04.020 Except in the case of the following drugs, every package of a drug that is a prescription drug shall carry the symbol “Pr” on the upper left quarter of the principal display panel of both its inner and outer labels or, in the case of a single dose container, on the upper left quarter of its outer label:
(a) a drug sold to a person who holds an establishment licence; and
(b) a drug sold under a prescription.
- SOR/80-543, s. 10
- SOR/97-12, s. 40
- SOR/2001-181, s. 4
- SOR/2013-122, s. 17
Bacterial Vaccines, Products Analogous to Bacterial Vaccines
C.04.050 Except as provided in this Division, a bacterial vaccine shall be a sterile suspension of killed cultures of bacteria, with or without the addition of other medication, and shall not include an autogenous vaccine.
C.04.051 No person shall sell a bacterial vaccine unless the culture that has been used in its preparation has been tested by an acceptable method for identity and purity and when so tested it shall be true to name and a pure strain, and a record of the culture shall be maintained which shall include a statement of its origin, properties and characteristics.
C.04.052 No fabricator shall use a substrate (culture medium), in the production of a bacterial vaccine, that contains any horse meat or horse serum.
- SOR/97-12, s. 61
C.04.053 A fabricator of a bacterial vaccine prepared from a bacterium that does not grow readily in ordinary culture media shall test its sterility in media which are specially favourable to the growth of such bacterium, and it shall be sterile.
- SOR/97-12, s. 61
C.04.054 Except as provided in sections C.04.083, C.04.084 and C.04.090, both the inner and outer labels of every multiple-dose container and the outer label of every single-dose container of a bacterial vaccine shall carry a statement of
(a) the number of bacteria per millilitre, or the weight of dried substance of bacteria per millilitre,
(b) the number of bacteria per millilitre, or the weight of dried substance of bacteria per millilitre, of each species or immunogenic type for a vaccine that contains a number of different species or immunogenic types of bacteria,
(c) the exact nature and amount of any substance, other than a simple diluent, combined with such vaccine, and
(d) the recommended dose,
and the inner label of a single-dose container shall carry a statement that it contains only one dose.
C.04.055 The expiration date of a bacterial vaccine shall be not later than 18 months after the date of manufacture or the date of issue.
Typhoid Vaccine
C.04.060 Cultures of Salmonella typhosa used in the preparation of typhoid vaccine shall be smooth, motile, and in the Vi form, with the following antigenic structure IX,XII,Vi; d.-.
C.04.061 No person shall sell any lot of typhoid vaccine unless such lot has been shown to meet a test for potency made by an acceptable method.
Pertussis Vaccine
C.04.065 A fabricator shall, in the preparation of pertussis (whooping cough) vaccine, use only strains of Bordetella pertussis that meet the requirements of an antigenic test made by an acceptable method.
- SOR/90-217, s. 1
- SOR/97-12, s. 61
C.04.066 No person shall sell any lot of pertussis (whooping cough) vaccine unless such lot has been shown to meet a test for potency made by an acceptable method.
B.C.G. (Bacille Calmette-Guerin) Vaccine
C.04.070 B.C.G. vaccine shall be prepared from living B.C.G. organisms that
(a) have been obtained directly from a source approved by the Minister;
(b) are proved to be non-pathogenic by an acceptable method; and
(c) have a history of successful use in the production of B.C.G. vaccine.
- SOR/2018-69, s. 27
C.04.071 No fabricator shall employ any person in the manufacture of B.C.G. vaccine unless such person
(a) has been and remains free from all forms of tuberculous infection,
(b) undergoes every six months a medical examination, that shall include an X-ray examination of the chest, for the presence of tuberculosis, such examination being made by a qualified, practising physician who shall sign a certificate of such person’s freedom from tuberculosis, and such certificate shall be kept on file and be available at all times, and
(c) resides in a household that is at all times free from active tuberculosis,
nor shall a fabricator employ such person in any other laboratory position.
- SOR/97-12, s. 61
C.04.072 The preparation, preservation and packaging/labelling of B.C.G. vaccine shall be conducted under the direct supervision of an experienced bacteriologist who has
(a) not less than three years postgraduate training in bacteriology and immunology;
(b) specialized in the field of bacteriology; and
(c) at least one year of practical experience in the manufacture of B.C.G. vaccine.
- SOR/97-12, s. 41
C.04.073 No fabricator shall permit any culture that is not a B.C.G. culture to be at any time on any premises that are used for the manufacture of B.C.G. vaccine.
- SOR/97-12, s. 61
C.04.074 A packager/labeller shall test by an acceptable method, after filling of the final container, each lot of B.C.G. vaccine for the presence of contaminating micro-organisms and when so tested it shall be free therefrom.
- SOR/97-12, s. 65
C.04.075 Notwithstanding section C.04.074, a fluid B.C.G. vaccine may be released for sale if no growth has appeared upon the test culture medium after an incubation of 24 hours, but if there is evidence of the presence of contaminating micro-organisms in any lot during the test period of 10 days the packager/labeller shall at once recall such lot.
- SOR/97-12, s. 65
C.04.076 Every fabricator and packager/labeller shall determine the number of viable B.C.G. organisms in each lot of vaccine by an acceptable method and shall keep a record of the number.
- SOR/97-12, s. 63
C.04.077 A fabricator of B.C.G. vaccine shall keep, at a temperature not exceeding 5.0°C, and for not less than six months,
(a) the culture on glycerine-water potato medium from which the Sauton I and Sauton II subcultures were made, and
(b) not less than six vials of the final product
from each lot thereof.
- SOR/97-12, s. 61
C.04.078 Every fabricator and packager/labeller of B.C.G. vaccine shall keep, in form satisfactory to the Minister, continuous clinical records of the use of B.C.G. vaccine in humans.
- SOR/97-12, s. 63
C.04.079 A fabricator of B.C.G. vaccine shall examine pathologically all test animals used and shall immediately report to the Minister any evidence of active, progressive tuberculosis in any such animals.
- SOR/97-12, s. 61
C.04.080 The expiration date for B.C.G. vaccine shall be not more than
(a) 10 days after harvesting in the case of fluid vaccine;
(b) 12 months after harvesting in the case of freeze dried vaccine stored at a temperature of 4°C or above; or
(c) 20 months after harvesting in the case of freeze dried vaccine stored at a temperature below 4°C.
C.04.081 No person shall sell fluid B.C.G. vaccine that is not packaged in containers sealed by fusion.
C.04.082 No inner label shall be required for fluid B.C.G. vaccine in single-dose containers.
C.04.083 The label of fluid B.C.G. vaccine shall carry, in lieu of the statements provided in paragraphs C.04.054(a) and (b), a statement of
(a) the weight of bacteria per millilitre; and
(b) the route of administration of the vaccine.
C.04.084 The label of freeze-dried B.C.G. vaccine shall carry, in lieu of the statements provided in paragraphs C.04.054(a) and (b), a statement of
(a) the amount of bacteria per vial or per dose; and
(b) the route of administration of the vaccine.
C.04.085 The provisions of subparagraph C.04.019(b)(iv) do not apply to freeze-dried B.C.G. vaccine.
Products Analogous to Bacterial Vaccines
C.04.090 A product analogous to a bacterial vaccine shall be
(a) a bacterial antigen, other than a bacterial vaccine, such as a lysate, or
(b) an extract prepared from a bacterial culture,
and shall conform to the requirements of these Regulations for bacterial vaccines except those of paragraphs (a) and (b) of C.04.054.
C.04.091 The expiration date of a product analogous to a bacterial vaccine shall be not later than 18 months after the date of manufacture or the date of issue, but for dried tuberculin and tuberculin containing at least 50 per cent glycerin the expiration date shall be not later than five years after the date of manufacture or the date of issue, and for all other tuberculins not more than 12 months after the date of manufacture or the date of issue.
Virus and Rickettsial Vaccines
C.04.100 A virus vaccine, rickettsial vaccine, shall be a suspension of, or prepared from, living or killed viruses or rickettsiae.
C.04.101 No person shall sell a virus or a rickettsial vaccine unless the fabricator has submitted to the Minister details of the source of the strains of viruses or rickettsiae used, the method of their propagation, the method of fabrication of the vaccine, the methods employed for determining sterility, safety, identity and potency and any other tests required by these Regulations.
- SOR/95-411, s. 2
- SOR/97-12, s. 42
C.04.102 Upon written request from the Minister every fabricator and packager/labeller shall submit with respect to each lot of virus or rickettsial vaccine, when ready for sale, detailed protocols of sterility, safety, identity, potency, and of any other tests required by these Regulations.
- SOR/97-12, s. 63
- SOR/2018-69, s. 27
Smallpox Vaccine
C.04.110 Smallpox vaccine
(a) shall be a virus vaccine;
(b) shall be the living virus of vaccinia or its derivatives obtained from
(i) the vesicles produced in the skin of healthy calves by inoculation of vaccinia virus,
(ii) specifically infected membranes of chick embryos, or
(iii) suitable tissue culture infected with vaccinia virus or its derivatives; and
(c) shall be in fluid or dried form.
- SOR/2006-2, s. 1
C.04.111 Every fabricator and packager/labeller shall fabricate and package/label smallpox vaccine only in an independent unit that is isolated from all other laboratory activities, and in or about which no extraneous materials are permitted or stored.
- SOR/97-12, s. 43
C.04.112 A fabricator shall exclude the personnel who care for the vaccine animals from horse stables and paddocks and from contact with horses while smallpox vaccine is being propagated.
- SOR/97-12, s. 61
C.04.113 Every fabricator and packager/labeller shall dispense smallpox vaccine only in sterile glass containers that are sealed under aseptic conditions.
- SOR/97-12, s. 63
C.04.114 Every fabricator and packager/labeller shall test smallpox vaccine to establish that it is free from
(a) spore-forming anaerobic micro-organisms;
(b) coagulase positive staphylococci;
(c) haemolytic streptococci; and
(d) any other contaminating pathogenic micro-organisms.
- SOR/97-12, s. 63
C.04.115 Smallpox vaccine, when tested by acceptable methods,
(a) shall be free from extraneous micro-organisms, in the case of vaccine prepared for use by jet gun; and
(b) shall contain not more than 500 viable non-pathogenic bacteria per millilitre, in the case of vaccine prepared for use by the multiple pressure technique or by scarification.
C.04.116 Smallpox vaccine must demonstrate evidence of disease prevention that is at least equivalent to that of a vaccine that
(a) is known to prevent human to human transmission of smallpox; and
(b) meets the potency of equal to or greater than 108 pockforming units per millilitre, as determined using chick embryo chorioallantoic membranes.
- SOR/2006-2, s. 2
C.04.117 No person shall sell smallpox vaccine unless
(a) in the case of fluid vaccine, it has been stored at a temperature below -10°C;
(b) in the case of dried vaccine, it has been stored at a temperature below 10°C; and
(c) the outer label carries a statement that it shall be stored at a temperature of not more than 5°C.
- SOR/97-12, s. 44
C.04.118 Notwithstanding the provisions of section C.04.003, the date of issue of smallpox vaccine shall be not later than
(a) in the case of fluid vaccine, nine months after the date of manufacture; and
(b) in the case of dried vaccine, 24 months after the date of manufacture.
C.04.119 The expiration date of smallpox vaccine shall not exceed the following, unless supported by evidence of stability satisfactory to the Minister:
(a) in the case of fluid vaccine, 3 months after the date of issue; or
(b) in the case of dried vaccine, 12 months after the date of issue.
- SOR/2006-2, s. 3
C.04.120 No inner label shall be required for smallpox vaccine in single-dose containers or when dispensed in capillary tubes.
C.04.121 No person shall sell smallpox vaccine to which an antibiotic has been added.
Poliomyelitis Vaccine
C.04.122 Poliomyelitis vaccine shall be an aqueous suspension of killed poliomyelitis viruses, Types I, II, and III.
C.04.123 Poliomyelitis vaccine shall be prepared in acceptable tissue culture medium from strains of poliomyelitis virus proven capable of producing vaccine of acceptable potency.
C.04.124 Poliomyelitis vaccine in its final form shall contain not more than 0.35 milligram per millilitre of total nitrogen, nor more than one part per million of animal serum.
C.04.125 No person shall sell poliomyelitis vaccine unless it has been tested by an acceptable method for potency and safety and when so tested it shall be safe and of acceptable potency.
C.04.126 The outer label shall carry a statement of any antibiotic present in the vaccine.
C.04.127 The expiration date of the poliomyelitis vaccine shall be not later than 12 months after the date of the last satisfactory potency test unless evidence, satisfactory to the Minister, is presented that a longer period is appropriate.
- SOR/85-715, s. 6
- SOR/2018-69, s. 27
Poliovirus Vaccine, Live, Oral
C.04.128 Poliovirus Vaccine, Live, Oral or Poliovirus Vaccine, Live, Oral (Naming the strains) shall be prepared from living poliomyelitis virus types I, II and III that
(a) have been obtained directly from a source acceptable to the Minister;
(b) are shown to be genetically stable by an acceptable method;
(c) are shown to be non-pathogenic when given orally to humans;
(d) are proved to be capable of multiplying in the human alimentary tract and of producing type specific neutralizing antibodies when administered orally; and
(e) have a history of successful use in the production of polio-virus vaccine, live, oral.
- SOR/2018-69, s. 27
C.04.129 Poliovirus vaccine, live, oral, shall be fabricated, packaged/labelled and tested in premises separated from buildings where other products are fabricated, packaged/labelled or tested, and from buildings where control tests involving the use of cell lines or virus strains not employed in the fabrication, packaging/labelling and testing of poliovirus vaccine, live, oral, are carried out.
- SOR/97-12, s. 45
C.04.130 No fabricator shall permit the introduction of any bacterial or viral cultures other than those used in the manufacture of poliovirus vaccine, live, oral on any premises that are used for the manufacture of poliovirus vaccine, live, oral.
- SOR/97-12, s. 61
C.04.131 Notwithstanding sections C.04.129 and C.04.130, a fabricator may manufacture other drugs in an area in which polio-virus vaccine, live, oral is manufactured at times when that vaccine is not being manufactured, if
(a) both prior to and following each manufacture the area is cleaned and disinfected by methods acceptable to the Minister; and
(b) the fabricator has received written permission from the Minister to carry out such manufacture.
- SOR/97-12, s. 61
- SOR/2018-69, s. 27
C.04.132 Poliovirus vaccine, live, oral shall be prepared only
(a) in a tissue culture,
(b) in a medium, and
(c) by methods
acceptable to the Minister.
- SOR/2018-69, s. 27
C.04.133 No fabricator shall sell poliovirus vaccine, live, oral, unless he has tested each lot for extraneous micro-organisms and the vaccine is free therefrom.
- SOR/97-12, s. 61
C.04.134 A fabricator of poliovirus vaccine, live, oral shall test, by a method acceptable to the Minister, each lot of vaccine for neurovirulence and for genetic markers and it shall meet the requirements established by the Minister.
- SOR/97-12, s. 61
- SOR/2018-69, s. 27
C.04.135 No fabricator shall employ any person in the manufacture of poliovirus vaccine, live, oral unless such person
(a) is free from infectious disease;
(b) has been vaccinated successfully against poliomyelitis by poliovirus vaccine, live, oral; and
(c) has been proved by periodic tests to be a non-carrier of poliomyelitis virus.
- SOR/97-12, s. 61
C.04.136 A fabricator of poliovirus vaccine, live, oral shall not permit the entry to a building in which the vaccine is manufactured of any person who
(a) is not directly concerned with the manufacturing processes; or
(b) has been working on the same day with experimental animals or with infectious agents.
- SOR/97-12, s. 61
Bacteriophage
C.04.137 Bacteriophage shall be a virus preparation with specific lytic action against micro-organisms actually or potentially pathogenic.
C.04.138 The expiration date of bacteriophage shall be not later than 12 months after the date of manufacture or the date of issue.
Toxins, Toxoids
Schick Test Reagents
C.04.140 Schick test reagents for the diagnosis of susceptibility to diphtheria shall be
(a) diphtheria toxin for Schick test;
(b) Schick control; and
(c) diphtheria toxin for Schick test with control.
C.04.141 Diphtheria toxin for Schick test shall be sterile diluted diphtheria toxin stabilized by an acceptable method.
C.04.142 Schick control shall be suitably diluted
(a) diphtheria toxoid; or
(b) sterile diphtheria toxin heated at a temperature of 95°C for five minutes.
C.04.143 The human test dose of diphtheria toxin for Schick test, when aged toxin containing a preservative is used, shall be determined by
(a) intracutaneous injection into normal guinea pigs in mixtures with different proportions of diphtheria antitoxin, and one test dose mixed with 1/750 or more of a unit of antitoxin must cause no local reaction but mixed with 1/1,250 or less of a unit of antitoxin must cause a definite local reaction of the type known as the “positive Schick reaction”; and
(b) intracutaneous injection into normal guinea pigs without admixture with antitoxin, and 1/50 of one test dose must not cause, and 1/25 of one test dose must cause, a definite local reaction of the type known as the “positive Schick reaction”.
C.04.144 The human test dose of diphtheria toxin for Schick test, when fresh toxin containing no preservative is used, shall be determined by
(a) intracutaneous injection into normal guinea pigs in mixtures with different proportions of diphtheria antitoxin, and one test dose mixed with 1/750 or more of a unit of antitoxin must cause no local reaction, but mixed with 1/1,500 or less of a unit of antitoxin must cause a definite local reaction of the type known as the “positive Schick reaction”; and
(b) intracutaneous injection into normal guinea pigs without admixture with antitoxin, and 1/100 of one test dose must not cause, and 1/50 of one test dose must cause, a definite local reaction of the type known as the “positive Schick reaction”.
C.04.145 The human test dose for the Schick control shall give a negative Schick reaction when injected intracutaneously into normal guinea pigs.
C.04.146 No person shall sell diphtheria toxin for Schick test unless both the inner and the outer labels carry a statement of the number of human test doses it contains together with the name of any stabilizer.
C.04.147 The expiration date of Schick test reagents for the diagnosis of susceptibility to diphtheria shall be not later than 12 months after the date of manufacture or the date of issue.
Diphtheria Toxoid
C.04.160 Liquid diphtheria toxoid shall be sterile, formalized, detoxified diphtheria toxin and shall not contain more than 0.02 per cent free formaldehyde.
C.04.161 Diphtheria toxoid alum precipitated shall be prepared from diphtheria toxoid, and shall not contain more than 15 milligrams of alum per human dose.
C.04.162 The alum used in the preparation of diphtheria toxoid alum precipitated shall contain not less than 99.5 per cent pure potassium alum, Al K(SO4)2,12H2O.
C.04.163 No fabricator shall use a culture medium for the production of diphtheria toxin that contains horse protein or Witte peptone or that has not been freed as far as possible from any other allergenic ingredient.
- SOR/97-12, s. 61
C.04.164 Diphtheria toxin from which diphtheria toxoid is prepared shall have a toxicity, as indicated by an L+dose, of not more than 0.20 millilitre or by an M.L.D. of not more than 0.0025 millilitre.
C.04.165 A fabricator shall test each bulk container of diphtheria toxoid, before being dispensed into the final containers, for toxicity by an acceptable method, and it shall be non-toxic.
- SOR/97-12, s. 61
C.04.166 No person shall sell any lot of diphtheria toxoid unless such lot has been shown to meet a test for antigenicity made by an acceptable method.
C.04.167 A fabricator shall fill diphtheria toxoid aseptically into clear glass containers and where preservative is not added shall seal the containers by fusion.
- SOR/97-12, s. 61
C.04.168 No person shall sell diphtheria toxoid that contains phenol.
C.04.169 No person shall sell diphtheria toxoid unless both the inner and the outer labels carry a statement of the appropriate dose for purposes of immunization.
C.04.170 The expiration date of diphtheria toxoid shall be not later than two years after the date of manufacture or the date of issue.
Tetanus Toxoid
C.04.180 Liquid tetanus toxoid shall be sterile, formalized, detoxified tetanus toxin, and shall not contain more than 0.02 per cent free formaldehyde.
C.04.181 Tetanus toxoid alum precipitated shall be prepared from tetanus toxoid, and shall not contain more than 15 milligrams of alum per human dose.
C.04.182 The alum used in the preparation of tetanus toxoid alum precipitated shall contain not less than 99.5 per cent pure potassium alum, Al K(SO4)2, 12H2O.
C.04.183 No fabricator shall use a culture medium for the production of tetanus toxin that contains horse protein or Witte peptone or that has not been freed as far as possible from any other allergenic ingredient.
- SOR/97-12, s. 61
C.04.184 Tetanus toxin from which tetanus toxoid is prepared shall have a toxicity as indicated by an M.L.D. for the guinea pig of not more than 0.0001 millilitre.
C.04.185 A packager/labeller shall test each bulk container of tetanus toxoid, before being dispensed into the final containers, for toxicity by an acceptable method, and it shall be non-toxic.
- SOR/97-12, s. 65
C.04.186 No person shall sell any lot of tetanus toxoid unless such lot has been shown to meet a test for antigenicity made by an acceptable method.
C.04.187 No person shall sell tetanus toxoid unless both the inner and the outer labels carry a statement of the appropriate dose for purposes of immunization.
C.04.188 A fabricator shall fill tetanus toxoid aseptically into clear glass containers and where a preservative is not added shall seal the container by fusion.
- SOR/97-12, s. 61
C.04.189 No person shall sell tetanus toxoid that contains phenol.
C.04.190 The expiration date of tetanus toxoid shall be not later than two years after the date of manufacture or the date of issue.
Antitoxins, Antisera
C.04.210 An antitoxin or antiserum shall be the serum or fraction thereof separated from the blood of animals that have been artificially immunized against the by-products or antigenic fractions of specific cultures of micro-organisms, or against specific venoms.
C.04.211 The potency of an antitoxin or antiserum shall be determined by an acceptable method and where applicable the unit of potency shall be the International Unit.
C.04.212 Liquid diphtheria antitoxin shall have a potency of not less than 500 International Units per millilitre.
C.04.213 Liquid tetanus antitoxin shall have a potency of not less than 400 International Units per millilitre.
C.04.214 A liquid antitoxin or antiserum shall contain not more than 20 per cent solids.
C.04.215 A dried antitoxin shall be prepared from a liquid antitoxin and, when reconstituted to the original volume of the liquid antitoxin, shall have a potency not less than that prescribed for such liquid antitoxin.
C.04.216 A dried antitoxin or antiserum shall not contain more than one per cent moisture when determined by an acceptable method.
C.04.217 Each lot of antitoxin or antiserum shall be tested by an acceptable method for pyrogenicity and it shall be pyrogen-free, and, after filling into the final containers, for identity and it shall be true to name.
C.04.218 No person shall sell an antitoxin or antiserum unless both the inner and the outer labels carry a statement of the species of animal used, when other than the horse, and the net contents in millilitres or the number of units in the container.
C.04.219 In respect of antitoxins, the expiration date shall be
(a) for liquid antitoxins with standards of potency, not later than five years after the date of manufacture;
(b) for dried antitoxins with standards of potency, not later than five years after the date of manufacture;
(c) for liquid antioxins with no standards of potency, not later than 12 months after the date of manufacture; and
(d) for dried antitoxins with no standards of potency, not later than five years after the date of manufacture.
C.04.220 In respect of antisera, the expiration date shall be
(a) for liquid antisera with standards of potency, not later than three years after the date of manufacture;
(b) for dried antisera with standards of potency, not later than five years after the date of manufacture;
(c) for liquid antisera with no standards of potency, not later than 12 months after the date of manufacture; and
(d) for dried antisera with no standards of potency, not later than five years after the date of manufacture.
Preparations from Human Sources
C.04.230 Preparations from human sources shall be pooled blood plasma, or pooled blood serum, or fractions of either separated by a method satisfactory to the Minister.
C.04.231 A fabricator shall obtain human serum, or human plasma, only from a person certified by a qualified medical practitioner to be healthy.
- SOR/97-12, s. 61
C.04.232 A fabricator shall not use a person to serve as a donor of blood, placenta, or cord who has a history of a disease transmissible by blood transfusion including syphilis, infectious hepatitis, or malaria.
- SOR/97-12, s. 61
C.04.233 The operation of drawing blood from a donor shall be under the supervision of a qualified medical practitioner, and shall be carried out in a suitable bleeding room under the control of the fabricator.
- SOR/97-12, s. 61
C.04.234 A fabricator shall obtain human placenta and cord used in the manufacture of preparations from human sources only from women confined in public hospitals, and the donor of such placenta and cord shall have been free from the toxaemias of pregnancy, and the placenta and cord shall not show gross evidence of any pathological condition.
- SOR/97-12, s. 61
C.04.235 (1) Subject to subsections (2) and (3), dried human serum, dried human plasma or dried fractions of either shall not contain more than one per cent moisture when determined by an acceptable method.
(2) Dried Rho(D) Immune Human globulin shall not contain more than three per cent moisture when determined by an acceptable method.
(3) Dried Antihemophilic Factor Human shall not contain more than two per cent moisture when determined by an acceptable method.
- SOR/81-334, s. 3
C.04.236 A fabricator shall provide directions or means for the removal of particles of such size as to be dangerous to the recipient from preparations from human sources that are issued in fluid form or that are reconstituted from the dried form.
- SOR/97-12, s. 61
C.04.237 A fabricator of preparations from human sources shall maintain complete records of all donors, which records shall include the medical certificate required by section C.04.231.
- SOR/97-12, s. 61
C.04.238 A fabricator, packager/labeller or distributor referred to in paragraph C.01A.003(b) may issue human serum or human plasma, or fractions of either of them, for prophylactic or therapeutic use in any of the following forms:
(a) immune human serum, which shall be serum separated from the blood of persons recovered from the disease or from persons specifically immunized against the disease for which the serum is intended as a prophylactic or therapeutic agent;
(b) immune human globulins, or other immune human serum fractions, which shall be prepared from immune human serum or plasma;
(c) normal human serum, or normal human plasma, or fractions of either of these prepared from the blood of normal individuals; and
(d) dried products prepared from any of these.
- SOR/97-12, s. 46
C.04.239 No person shall sell a preparation from human sources unless both the inner and the outer labels clearly indicate that the preparation is derived from human sources.
C.04.240 The expiration date for preparations from human sources issued in fluid or dried form shall be not later than five years after the date of filling the immediate container.
C.04.241 The date of manufacture of preparations from human sources shall be the date of bleeding the donor.
C.04.300 and C.04.301 [Repealed, SOR/81-335, s. 3]
C.04.400 [Repealed, SOR/2013-179, s. 4]
C.04.401 [Repealed, SOR/2013-179, s. 4]
C.04.402 [Repealed, SOR/2013-179, s. 4]
C.04.403 [Repealed, SOR/2013-179, s. 4]
C.04.404 [Repealed, SOR/2013-179, s. 4]
C.04.405 [Repealed, SOR/2013-179, s. 4]
C.04.406 [Repealed, SOR/2013-179, s. 4]
C.04.407 [Repealed, SOR/2013-179, s. 4]
C.04.408 [Repealed, SOR/2013-179, s. 4]
C.04.409 [Repealed, SOR/2013-179, s. 4]
C.04.410 [Repealed, SOR/2013-179, s. 4]
C.04.411 [Repealed, SOR/2013-179, s. 4]
C.04.412 [Repealed, SOR/2013-179, s. 4]
C.04.413 [Repealed, SOR/2013-179, s. 4]
C.04.414 [Repealed, SOR/2013-179, s. 4]
C.04.415 [Repealed, SOR/2013-179, s. 4]
C.04.416 [Repealed, SOR/2013-179, s. 4]
C.04.417 [Repealed, SOR/2013-179, s. 4]
C.04.418 [Repealed, SOR/2013-179, s. 4]
C.04.419 [Repealed, SOR/2013-179, s. 4]
C.04.420 [Repealed, SOR/2013-179, s. 4]
C.04.421 [Repealed, SOR/2013-179, s. 4]
C.04.422 [Repealed, SOR/2013-179, s. 4]
C.04.423 [Repealed, SOR/2013-179, s. 4]
C.04.424 [Repealed, SOR/2006-353, s. 1]
C.04.425 [Repealed, SOR/2006-353, s. 1]
C.04.426 [Repealed, SOR/2006-353, s. 1]
C.04.427 [Repealed, SOR/97-12, s. 50]
C.04.428 [Repealed, SOR/2006-353, s. 1]
Insulin Preparations
- SOR/82-769, s. 5
C.04.550 (1) Insulin means the active principle of the pancreas that affects the metabolism of carbohydrates in the animal body and that is of value in the treatment of diabetes mellitus.
(2) The Canadian Reference Standard for insulin shall be the International Standard therefor.
(3) The insulin preparations described in these Regulations shall contain insulin to which may be added only such ingredients as are prescribed in these Regulations.
(4) The potency of an insulin preparation shall be expressed in units per cubic centimetre and each unit per cubic centimetre shall provide one International Unit of insulin per cubic centimetre.
- SOR/82-769, s. 4
C.04.551 No person shall sell or dispense an insulin preparation that has not been stored by him continuously at a temperature between 35° and 50°F (2° and 10°C).
- SOR/82-769, s. 4
C.04.552 The zinc-insulin crystals used in an insulin preparation shall contain, as determined by an acceptable method,
(a) not less than 21 International Units of insulin per milligram, and
(b) on the dry basis, not less than 0.30 per cent and not more than 0.90 per cent zinc.
- SOR/82-769, s. 4
Insulin Injection or Insulin
C.04.553 The insulin preparation, “Insulin injection” or “Insulin” shall be a clear colourless or almost colourless sterile solution free from turbidity and insoluble matter, prepared from insulin or zinc insulin crystals, shall have a pH of not less than 2.5 or more than 3.5, or not less than 7.0 or more than 7.8 and shall contain
(a) weight by volume,
(i) not less than 0.1 per cent and not more than 0.25 per cent of either phenol or cresol, and
(ii) not less than 1.4 per cent and not more than 1.8 per cent glycerin; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 7.0 milligrams of nitrogen for Insulin Injection prepared from zinc-insulin crystals, and not more than 8.5 milligrams of nitrogen for Insulin Injection other than that made from zinc-insulin crystals,
(ii) not less than 0.10 milligram and not more than 0.40 milligram of zinc for Insulin Injection prepared from zinc-insulin crystals, and not more than 0.40 milligram of zinc for Insulin Injection other than that made from zinc-insulin crystals, and
(iii) in the case of Insulin Injection other than that made from zinc-insulin crystals, not more than 1.0 milligram of ash.
- SOR/82-769, s. 4
- SOR/85-715, s. 7
C.04.554 No person shall sell Insulin Injection unless,
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres;
(b) the vial label indicates that each cubic centimetre has a potency equal to
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin; and
(c) each cubic centimetre thereof has an actual potency that is at least 95 per cent and does not exceed 105 per cent of the potency indicated on the label as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.555 (1) A fabricator shall not sell Insulin Injection unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Insulin Injection
(i) protocols of assay of its potency expressed in International Units per cubic centimetre, in the case of insulin, and in International Units per milligram, in the case of zinc-insulin crystals,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals,
(iii) a report of the ash content in the case of insulin, and
(iv) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the first finished lot of Insulin Injection prepared from each master lot of insulin or zinc-insulin crystals, a report on the amount of each component thereof; and
(c) for the first filling of the first finished lot of Insulin Injection from each master lot of insulin or zinc-insulin crystals,
(i) a report of assay of its nitrogen content in milligrams per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per 1,000 International Units of insulin, and
(iii) a report on the determination of its pH.
(iv) [Repealed, SOR/95-203, s. 2]
- SOR/82-769, s. 4
- SOR/95-203, s. 2
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.556 The expiration date printed on the inner and outer labels of every package of Insulin Injection shall be a date not later than two years after the date of removal for distribution from the fabricator’s place of storage.
- SOR/82-769, s. 4
- SOR/97-12, s. 61
Insulin Zinc Suspension — Rapid
C.04.557 The insulin preparation “Insulin Zinc Suspension — Rapid” shall be a sterile suspension in a buffered aqueous medium, of insulin modified by the addition of zinc in such a way that the suspended precipitate consists of amorphous material, shall have a pH of not less than 7.0 and not more than 7.8 and shall contain,
(a) weight by volume,
(i) not less than 0.15 per cent and not more than 0.17 per cent of sodium acetate (NaC2H3O2.3H2O),
(ii) not less than 0.65 per cent and not more than 0.75 per cent of sodium chloride, and
(iii) not less than 0.09 per cent and not more than 0.11 per cent of methyl-p-hydroxybenzoate; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 7.0 milligrams of nitrogen; and
(ii) not less than 1.2 milligrams and not more than 2.5 milligrams of zinc, of which not less than 20 per cent and not more than 65 per cent shall be in the supernatant liquid.
- SOR/80-545, s. 1
- SOR/82-769, s. 4
- SOR/85-715, s. 8
C.04.558 The insulin used in the preparation of Insulin Zinc Suspension — Rapid shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40, 80 or 100 International Units of insulin in each cubic centimetre of Insulin Zinc Suspension-Rapid when the precipitate is suspended uniformly.
- SOR/82-769, s. 4
C.04.559 The clear supernatant liquid obtained from Insulin Zinc Suspension — Rapid shall contain not more than 1.0 International Unit of Insulin per cubic centimetre when the potency of the insulin preparation is 40 units per cubic centimetre, and not more than 1.5 International Units of insulin per cubic centimetre when the potency of the insulin preparation is either 80 units or 100 units per cubic centimetre, as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.560 No person shall sell Insulin Zinc Suspension — Rapid unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides, when the precipitate is suspended uniformly,
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin.
- SOR/82-769, s. 4
C.04.561 (1) A fabricator shall not sell Insulin Zinc Suspension — Rapid unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Insulin Zinc Suspension — Rapid,
(i) protocols of assay of its potency expressed in International Units per cubic centimetre in the case of insulin, and in International Units per milligram in the case of zinc-insulin crystals,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals, and
(iii) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the first finished lot of Insulin Zinc Suspension — Rapid prepared from each master lot of insulin or zinc-insulin crystals
(i) a report on the amount of each component used in the preparation,
(ii) a report of assay of its nitrogen content per 1,000 International Units of insulin,
(iii) a report of assay of its zinc content per 1,000 International Units of insulin,
(iv) a report of the insulin content in International Units per cubic centimetre of the supernatant liquid after removal of the suspended precipitate,
(v) a report of assay of the zinc content of the supernatant liquid after removal of the suspended precipitate,
(vi) a report on the determination of its pH, and
(vii) a report on the microscopic appearance of the suspended precipitate; and
(c) for the first filling of the first finished lot of Insulin Zinc Suspension — Rapid from each master lot of insulin or zinc-insulin crystals,
(i) a report on the determination of its pH,
(ii) a report on the microscopic examination of the precipitate, and
(iii) a report on its identification, as determined by an acceptable method.
(iv) [Repealed, SOR/95-203, s. 3]
- SOR/82-769, s. 4
- SOR/95-203, s. 3
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.562 The expiration date printed on the inner and outer labels of every package of Insulin Zinc Suspension — Rapid shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
Insulin Zinc Suspension — Medium
C.04.563 The insulin preparation “Insulin Zinc Suspension — Medium” shall be a sterile suspension, in a buffered aqueous medium, of insulin modified by the addition of zinc in such a way that the suspended precipitate consists of a mixture of crystals and amorphous material in an approximate ratio of seven parts of crystals to three parts of amorphous material, shall have a pH of not less than 7.0 and not more than 7.8 and shall contain,
(a) weight by volume,
(i) not less than 0.15 per cent and not more than 0.17 per cent of sodium acetate (NaC2H3O2.3H2O),
(ii) not less than 0.65 per cent and not more than 0.75 per cent of sodium chloride, and
(iii) not less than 0.09 per cent and not more than 0.11 per cent of methyl-p-hydroxybenzoate; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 7.0 milligrams of nitrogen of which not less than 63 per cent and not more than 73 per cent shall be in the crystalline component, and
(ii) not less than 1.2 milligrams and not more than 2.5 milligrams of zinc, of which not less than 20 per cent and not more than 65 per cent shall be in the supernatant liquid.
- SOR/80-545, s. 2
- SOR/82-769, s. 4
- SOR/85-715, s. 9
- SOR/88-323, s. 7
C.04.564 The insulin used in the preparation of Insulin Zinc Suspension — Medium shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40, 80 or 100 International Units of insulin in each cubic centimetre of the preparation when the precipitate is suspended uniformly.
- SOR/82-769, s. 4
C.04.565 The clear supernatant liquid obtained from Insulin Zinc Suspension — Medium shall contain not more than 1.0 International Unit of insulin per cubic centimetre when the potency of the insulin preparation is 40 units per cubic centimetre, and not more than 1.5 International Units of insulin per cubic centimetre when the potency of the insulin preparation is either 80 units or 100 units per cubic centimetre, as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.566 No person shall sell Insulin Zinc Suspension-Medium unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides, when the precipitate is suspended uniformly,
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin.
- SOR/82-769, s. 4
C.04.567 (1) A fabricator shall not sell Insulin Zinc Suspension-Medium unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Insulin Zinc Suspension-Medium,
(i) protocols of assay of its potency expressed in International Units per cubic centimetre in the case of insulin, and in International Units per milligram in the case of zinc-insulin crystals,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals, and
(iii) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the first finished lot of Insulin Zinc Suspension-Medium prepared from each master lot of insulin or zinc-insulin crystals,
(i) a report on the amount of each component used in the preparation,
(ii) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iv) a report of the insulin content, in International Units per cubic centimetre, of the supernatant liquid after removal of the suspended precipitate,
(v) a report on the determination of the proportion of the nitrogen in the crystalline component of the suspended precipitate,
(vi) a report of assay of the zinc content of the supernatant liquid after removal of the suspended precipitate,
(vii) a report on the determination of its pH, and
(viii) a report on the microscopic appearance of the suspended precipitate; and
(c) for the first filling of the first finished lot of Insulin Zinc Suspension — Medium from each master lot of insulin or zinc-insulin crystals,
(i) a report on the determination of its pH,
(ii) a report on the microscopic examination of the precipitate, and
(iii) a report on its identification as determined by an acceptable method.
(iv) [Repealed, SOR/95-203, s. 4]
- SOR/82-769, s. 4
- SOR/95-203, s. 4
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.568 The expiration date printed on the inner and outer labels of Insulin Zinc Suspension — Medium shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
Insulin Zinc Suspension — Prolonged
C.04.569 The insulin preparation “Insulin Zinc Suspension — Prolonged” shall be a sterile suspension in a buffered aqueous medium of insulin modified by the addition of zinc in such a way that the suspended precipitate consists of crystals with not more than a trace of amorphous material, shall have a pH of not less than 7.0 and not more than 7.8 and shall contain
(a) weight by volume,
(i) not less than 0.15 per cent and not more than 0.17 per cent of sodium acetate (NaC2H3O2.3H2O),
(ii) not less than 0.65 per cent and not more than 0.75 per cent of sodium chloride, and
(iii) not less than 0.09 per cent and not more than 0.11 per cent of methyl-p-hydroxybenzoate; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 7.0 milligrams of nitrogen, of which not less than 90 per cent shall be in the crystalline component, and
(ii) not less than 1.2 milligrams and not more than 2.5 milligrams of zinc, of which not less than 20 per cent and not more than 65 per cent shall be in the supernatant liquid.
- SOR/80-545, s. 3
- SOR/82-769, s. 4
- SOR/85-715, s. 10
C.04.570 The insulin used in the preparation of Insulin Zinc Suspension — Prolonged shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40, 80 or 100 International Units of insulin in each cubic centimetre of the preparation when the precipitate is suspended uniformly.
- SOR/82-769, s. 4
C.04.571 The clear supernatant liquid obtained from Insulin Zinc Suspension — Prolonged shall contain not more than 1.0 International Unit of insulin per cubic centimetre when the potency of the insulin preparation is 40 units per cubic centimetre, and not more than 1.5 International Units of insulin per cubic centimetre when the potency of the insulin preparation is either 80 units or 100 units per cubic centimetre, as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.572 No person shall sell Insulin Zinc Suspension — Prolonged unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides, when the precipitate is suspended uniformly,
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin.
- SOR/82-769, s. 4
C.04.573 (1) A fabricator shall not sell Insulin Zinc Suspension — Prolonged unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Insulin Zinc Suspension — Prolonged,
(i) protocols of assay of its potency expressed in International Units per cubic centimetre in the case of insulin, and in International Units per milligram in the case of zinc-insulin crystals,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals, and
(iii) reports of assay of the nitrogen content in milligrams and of its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the first finished lot of Insulin Zinc Suspension — Prolonged prepared from each master lot of insulin or zinc-insulin crystals,
(i) a report on the amount of each component used in the preparation,
(ii) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iv) a report of the insulin content, in International Units per cubic centimetre, of the supernatant liquid after removal of the suspended precipitate,
(v) a report of the determination of the proportion of the nitrogen in the crystalline component of the suspended precipitate,
(vi) a report of assay of the zinc content of the supernatant liquid after removal of the suspended precipitate,
(vii) a report on the determination of its pH, and
(viii) a report on the microscopic appearance of the suspended precipitate; and
(c) for the first filling of the first finished lot of Insulin Zinc Suspension — Prolonged from each master lot of insulin or zinc-insulin crystals,
(i) a report on the determination of its pH,
(ii) a report on the microscopic examination of the precipitate, and
(iii) a report on its identification as determined by an acceptable method.
(iv) [Repealed, SOR/95-203, s. 5]
- SOR/82-769, s. 4
- SOR/95-203, s. 5
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.574 The expiration date printed on the inner and outer labels of every package of Insulin Zinc Suspension — Prolonged shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
Globin Insulin with Zinc
C.04.575 The insulin preparation “Globin Insulin with Zinc” shall be a sterile solution of insulin modified by the addition of globin prepared from beef blood, in the form of globin hydrochloride, and zinc, shall be a clear, yellowish, or almost colourless liquid free from insoluble matter and acceptably free from turbidity, shall have a pH of not less than 3.4 and not more than 3.8 and shall contain,
(a) weight by volume, not less than 1.3 per cent and not more than 1.7 per cent glycerin, and either
(i) not less than 0.15 per cent and not more than 0.20 per cent cresol, or
(ii) not less than 0.20 per cent and not more than 0.26 per cent phenol, and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 15.0 milligrams of total nitrogen,
(ii) not less than 36.0 milligrams and not more than 40.0 milligrams of globin calculated as 6.0 times the nitrogen content of the globin, and
(iii) not less than 2.5 milligrams and not more than 3.5 milligrams of zinc.
- SOR/82-769, s. 4
C.04.576 The globin hydrochloride used in the preparation of Globin Insulin with Zinc shall contain not less than 16.0 per cent and not more than 17.5 per cent nitrogen calculated on a dry, ash-free and hydrochloric acid-free basis, and its ash content shall be not more than 0.3 per cent as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.577 The insulin used in the preparation of Globin Insulin with Zinc shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40 or 80 International Units of insulin in each cubic centimetre of the Globin Insulin with Zinc.
- SOR/82-769, s. 4
C.04.578 (1) The Canadian Reference Standard for Globin Insulin with Zinc shall be the standard adopted therefor by the Minister from time to time.
(2) Upon application of a person who holds an establishment licence, the Minister shall furnish him with a portion of the Canadian Reference Standard with directions for comparative testing.
(3) The testing of the biological reaction of Globin Insulin with Zinc shall be made by an acceptable method and that biological reaction shall be comparable to the biological reaction of the portion of the Canadian Reference Standard furnished by the Minister.
- SOR/82-769, s. 4
- SOR/97-12, s. 64
- SOR/2018-69, ss. 31(E), 32(F)
C.04.579 No person shall sell Globin Insulin with Zinc unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides,
(i) 40 International Units of insulin, or
(ii) 80 International Units of insulin.
- SOR/82-769, s. 4
C.04.580 (1) A fabricator shall not sell Globin Insulin with Zinc unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Globin Insulin with Zinc,
(i) protocols of assay of its potency expressed in International Units per cubic centimetre in the case of insulin, and in International Units per milligram in the case of zinc-insulin crystals,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals, and
(iii) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the master lot of globin hydrochloride used in the preparation of Globin Insulin with Zinc, reports of assay of
(i) its nitrogen content in per cent calculated on a dry, ash-free and hydrochloric acid free basis,
(ii) its chloride content in per cent calculated as hydrochloride, and
(iii) its ash content in percentage;
(c) for the components used in the preparation of the trial mixture of Globin Insulin with Zinc, a report on the quantity of
(i) insulin in grams, or in International Units,
(ii) zinc in grams, or in milligrams, per 1,000 International Units of insulin,
(iii) globin hydrochloride in grams or in milligrams, per 1,000 International Units of insulin, and
(iv) the volume of the preparation in cubic centimetres or litres;
(d) for the trial mixture of Globin Insulin with Zinc,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iii) protocols of the biological reaction showing the retardation of the insulin effect, and
(iv) a report on the determination of its pH;
(e) for the first finished lot of Globin Insulin with Zinc from each trial mixture of Globin Insulin with Zinc, a report on the amount of each component in the preparation; and
(f) for the first filling of the first finished lot of Globin Insulin with Zinc from each trial mixture of Globin Insulin with Zinc,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin, and
(iii) a report on the determination of its pH.
(iv) [Repealed, SOR/95-203, s. 6]
- SOR/82-769, s. 4
- SOR/95-203, s. 6
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.581 The expiration date printed on the inner and outer labels of every package of Globin Insulin with Zinc shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
NPH Insulin or Isophane Insulin
C.04.582 The insulin preparation “NPH Insulin” or “Isophane Insulin” shall be a sterile preparation of rod-shaped crystals containing insulin, protamine and zinc, suspended in a buffered aqueous medium, shall have a pH of not less than 7.0 and not more than 7.8 and shall contain
(a) weight by volume, not less than 0.15 per cent and not more than 0.25 per cent anhydrous disodium phosphate, and either
(i) not less than 1.4 per cent and not more than 1.8 per cent glycerin and not less than 0.15 per cent and not more than 0.17 per cent metacresol and not less than 0.06 and not more than 0.07 per cent phenol, or
(ii) not less than 0.40 per cent and not more than 0.45 per cent sodium chloride and not less than 0.7 per cent and not more than 0.9 per cent glycerin and not less than 0.18 per cent and not more than 0.22 per cent metacresol; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 8.5 milligrams of nitrogen,
(ii) not less than 3.0 milligrams and not more than 6.0 milligrams of protamine except that the ratio of the protamine to the insulin shall be not less than the isophane ratio and shall not exceed the isophane ratio by more than 10 per cent,
(iii) not less than 0.16 milligram and not more than 0.40 milligram of zinc, and
(iv) no protease activity significant for the stability of NPH insulin.
- SOR/82-769, s. 4
- SOR/85-715, s. 11
C.04.583 The protamine used in preparing NPH Insulin shall be obtained from the sperm or from the mature testes of fish belonging to the family Salmonidae, genera Oncorhynchus Suckley, or Salmo Linne.
- SOR/82-769, s. 4
C.04.584 The isophane ratio means the minimum number of milligrams of protamine required to precipitate 100 International Units of insulin and shall be determined by an acceptable method.
- SOR/82-769, s. 4
C.04.585 The insulin used in the preparation of NPH Insulin shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40, 80 or 100 International Units of insulin in each cubic centimetre of the preparation when the precipitate is suspended uniformly.
- SOR/82-769, s. 4
C.04.586 The clear supernatant liquid obtained from NPH insulin shall contain not more than 0.4 International Units of insulin per cubic centimetre when the potency of the insulin preparation is 40 units per cubic centimetre, not more than 0.6 International Units of insulin per cubic centimetre when the potency of the insulin preparation is 80 units per cubic centimetre and not more than 0.7 International Units of insulin per cubic centimetre when the potency of the insulin preparation is 100 units per cubic centimetre, as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.587 No person shall sell NPH Insulin unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides,
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin.
- SOR/82-769, s. 4
C.04.588 (1) A fabricator shall not sell NPH Insulin unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of zinc-insulin crystals employed in the manufacture of NPH Insulin,
(i) protocols of assay of its potency in International Units per milligram,
(ii) a report of its moisture content in per cent determined by drying to constant weight at 100°C, and
(iii) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the master lot of protamine, a report of the isophane ratio for the insulin used in the preparation of the NPH Insulin;
(c) for the trial mixture of NPH Insulin,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iii) a report of the insulin content in International Units per cubic centimetre of the supernatant liquid after removal of the suspended precipitate,
(iv) a report on the determination of its pH, and
(v) a report on the microscopic examination of the precipitate;
(d) for the first finished lot of NPH Insulin from each trial mixture of NPH Insulin, a report on the amount of each component in the preparation; and
(e) for the first filling of the first finished lot of NPH Insulin from each trial mixture of NPH Insulin,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(iii) a report on the determination of its pH,
(iv) a report on the microscopic examination of the precipitate, and
(v) a report of its identification as determined by an acceptable method.
(vi) [Repealed, SOR/95-203, s. 7]
- SOR/82-769, s. 4
- SOR/95-203, s. 7
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.589 The expiration date printed on the inner and outer labels of NPH Insulin shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
Protamine Zinc Insulin
C.04.590 The insulin preparation “Protamine Zinc Insulin” shall be a sterile white suspension in a buffered aqueous medium, containing insulin modified by the addition of protamine and zinc, shall have a pH of not less than 7.1 and not more than 7.4, and shall contain,
(a) weight by volume,
(i) not less than 0.15 per cent and not more than 0.25 per cent anhydrous disodium phosphate,
(ii) not less than 1.4 per cent and not more than 1.8 per cent glycerin, and
(iii) either not less than 0.18 per cent and not more than 0.22 per cent cresol, or not less than 0.22 per cent and not more than 0.28 per cent phenol; and
(b) as determined by an acceptable method, for each 1,000 International Units of insulin,
(i) not more than 12.5 milligrams of total nitrogen,
(ii) not less than 10.0 milligrams and not more than 15.0 milligrams of protamine,
(iii) not less than 1.7 milligrams and not more than 2.5 milligrams of zinc.
- SOR/82-769, s. 4
C.04.591 The protamine used in the preparation of Protamine Zinc Insulin shall be obtained from the sperm or from the mature testes of fish belonging to the family Salmonidae, genera Oncorhunchus Suckley or Salmo Linne.
- SOR/82-769, s. 4
C.04.592 The insulin used in the preparation of Protamine Zinc Insulin shall be obtained from one or more master lots and shall be present in an amount sufficient to provide either 40, 80 or 100 International Units of insulin in each cubic centimetre of the preparation when the precipitate is suspended uniformly.
- SOR/82-769, s. 4
C.04.593 (1) The Canadian Reference Standard for Protamine Zinc Insulin shall be the standard adopted therefor by the Minister from time to time.
(2) Upon application of a person who holds an establishment licence, the Minister shall furnish him with a portion of the Canadian Reference Standard with directions for comparative testing.
(3) The testing of the biological reaction of Protamine Zinc Insulin shall be made by an acceptable method and that biological reaction shall be comparable to the biological reaction of the portion of the Canadian Reference Standard furnished by the Minister.
- SOR/82-769, s. 4
- SOR/97-12, s. 64
- SOR/2018-69, ss. 31(E), 32(F)
C.04.594 No person shall sell Protamine Zinc Insulin unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres; and
(b) each cubic centimetre thereof provides
(i) 40 International Units of insulin,
(ii) 80 International Units of insulin, or
(iii) 100 International Units of insulin.
- SOR/82-769, s. 4
C.04.595 (1) A fabricator shall not sell Protamine Zinc Insulin unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of insulin or zinc-insulin crystals employed in the manufacture of Protamine Zinc Insulin,
(i) protocols of assay of its potency in International Units per cubic centimetre in the case of insulin and in International Units per milligram in the case of zinc-insulin crystals,
(ii) a report on its moisture content in percentage determined by drying to constant weight at 100°C in the case of zinc-insulin crystals, and
(iii) reports of assay of its nitrogen content in milligrams, and its zinc content in milligrams per 1,000 International Units of insulin;
(b) for the components used in the preparation of the trial mixture of Protamine Zinc Insulin, a report on the quantity of
(i) insulin in grams or in International Units,
(ii) zinc in grams or in milligrams, per 1,000 International Units of insulin,
(iii) protamine in grams or in milligrams, per 1,000 International Units of insulin, and
(iv) the volume of the preparation in cubic centimetres or litres;
(c) for the trial mixture of Protamine Zinc Insulin,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units of insulin,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre per 1,000 International Units of insulin,
(iii) protocols of its biological reaction showing retardation of the insulin effect, and
(iv) a report on the determination of its pH;
(d) for the first finished lot of Protamine Zinc Insulin from each trial mixture of Protamine Zinc Insulin, a report on the amount of each component in the preparation; and
(e) for the first filling of the first finished lot of Protamine Zinc Insulin from each trial mixture of Protamine Zinc Insulin,
(i) a report of assay of its nitrogen content in milligrams per cubic centimetre or per 1,000 International Units,
(ii) a report of assay of its zinc content in milligrams per cubic centimetre or per 1,000 International Units, and
(iii) a report on the determination of its pH.
(iv) [Repealed, SOR/95-203, s. 8]
- SOR/82-769, s. 4
- SOR/95-203, s. 8
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.596 The expiration date printed on the inner and outer labels of every package of Protamine Zinc Insulin shall be a date not later than two years after the date of filling of the immediate container.
- SOR/82-769, s. 4
Sulphated Insulin
C.04.597 The insulin preparation “Sulphated Insulin” shall be a clear or slightly turbid, colourless or almost colourless, sterile, isotonic preparation of zinc-insulin crystals chemically modified by treatment with sulphuric acid, shall have a pH of not less than 6.0 and not more than 7.0, and shall contain,
(a) weight by volume,
(i) not less than 0.6 per cent and not more than 1.0 per cent sodium chloride, and
(ii) not less than 0.2 per cent and not more than 0.3 per cent phenol; and
(b) as determined by an acceptable method,
(i) not more than 200 milligrams protein for each 1,000 International Units of insulin, and
(ii) not less than 5.5 and not more than 6.5 sulphate groups per insulin molecule.
- SOR/82-769, s. 4
C.04.598 The neutralization ratio means the amount of anti-beef-insulin serum required to neutralize one unit of Sulphated Insulin divided by the amount required to neutralize one unit of beef insulin, and shall be determined by an acceptable method.
- SOR/82-769, s. 4
C.04.599 The neutralization ratio of Sulphated Insulin shall be not less than 4 to 1.
- SOR/82-769, s. 4
C.04.600 No person shall sell Sulphated Insulin unless
(a) it is dispensed in a vial of approximately 10 cubic centimetre capacity that contains an excess volume sufficient to permit withdrawal of 10 cubic centimetres, and
(b) each cubic centimetre thereof provides 100 International Units of insulin as determined by an acceptable method.
- SOR/82-769, s. 4
C.04.601 (1) A fabricator shall not sell Sulphated Insulin unless he
(a) has filed with the Minister, in accordance with subsection (2), a submission relating to that preparation, in a form and having a content satisfactory to the Minister;
(b) has furnished the Minister with such additional information as the Minister may require; and
(c) has received from the Minister a notice that the information contained in the submission is in accordance with the requirements of this section.
(2) A submission filed pursuant to subsection (1) shall include at least,
(a) for each master lot of zinc-insulin crystals employed in the manufacture of Sulphated Insulin,
(i) protocols of assay of its potency in International Units per milligram,
(ii) a report of its moisture content in percentage determined by drying to constant weight at 100°C, and
(iii) reports of assay of its nitrogen content in milligrams and its zinc content in milligrams per 1,000 International Units of insulin; and
(b) for each lot of Sulphated Insulin prepared from each master lot of zinc-insulin crystals,
(i) a report of the amount of each component,
(ii) a report of the protein content in milligrams per 1,000 International Units of insulin,
(iii) a report on the determination of the neutralization ratio,
(iv) a report on the determination of the number of sulphate groups per insulin molecule,
(v) protocols of assay of its potency expressed as International Units per cubic centimetre, and
(vi) a report on the determination of its pH.
(vii) [Repealed, SOR/95-203, s. 9]
- SOR/82-769, s. 4
- SOR/95-203, s. 9
- SOR/97-12, s. 61
- SOR/2018-69, ss. 27, 31(E), 32(F)
C.04.602 The expiration date printed on the inner and outer labels of every package of Sulphated Insulin shall be a date not later than two years after the date of filling of the immediate container.
- SOR/80-545, s. 4
- SOR/82-769, s. 4
Labelling of Insulin Preparations
- SOR/82-769, s. 8
C.04.650 The packager/labeller of Insulin Injection may label that insulin preparation “Insulin made from Zinc-Insulin crystals” only when it has been prepared from zinc-insulin crystals.
- SOR/82-769, s. 7
- SOR/97-12, s. 65
C.04.651 The packager/labeller of an insulin preparation shall print the information required by these Regulations to appear on both the inner and outer labels of every package of that insulin preparation as set out in the Table to this section.
TABLE
| Item | Column I | Column II | Column III |
|---|---|---|---|
| Insulin Preparation | Potency of Preparation | Special Printing Requirements for Label | |
| 1 | Insulin Injection, not labelled as set out in item 2. | (a) 40 units per cc. | (a) black ink on yellow stock. |
| (b) 80 units per cc. | (b) black ink on green stock. | ||
| (c) 100 units per cc. | (c) black ink on white stock. | ||
| 2 | Insulin Injection, labelled “Insulin made from Zinc-Insulin crystals.” | (a) 40 units per cc. | (a) red ink on grey stock. |
| (b) 80 units per cc. | (b) green ink on grey stock. | ||
| (c) 100 units per cc. | (c) black ink on white stock. | ||
| 3 | Insulin Zinc Suspension — Rapid, Insulin Zinc Suspension — Medium and Insulin Zinc Suspension — Prolonged. | (a) 40 units per cc. | (a) red ink on lavender stock plus a distinguishing mark or design. |
| (b) 80 units per cc. | (b) green ink on lavender stock plus a distinguishing mark or design. | ||
| (c) 100 units per cc. | (c) black ink on white stock. | ||
| 4 | Globin Insulin with Zinc. | (a) 40 units per cc. | (a) red ink on brown stock except that the expression “40 units per cubic centimetre” may be printed in white letters on a red background. |
| (b) 80 units per cc. | (b) green ink on brown stock except that the expression “80 units per cubic centimetre” may be printed in white letters on a green background. | ||
| 5 | NPH Insulin. | (a) 40 units per cc. | (a) red ink on blue stocks. |
| (b) 80 units per cc. | (b) green ink on blue stock. | ||
| (c) 100 units per cc. | (c) black ink on white stock. | ||
| 6 | Protamine Zinc Insulin. | (a) 40 units per cc. | (a) red ink on white stock. |
| (b) 80 units per cc. | (b) green ink on white stock. | ||
| (c) 100 units per cc. | (c) black ink on white stock | ||
| 7 | Sulphated Insulin. | 100 units per cc. | black ink on white stock plus the statement “Warning... Not for Ordinary Use... See Package Leaflet”. |
- SOR/82-769, s. 7
- SOR/97-12, s. 65
C.04.652 The packager/labeller of an insulin preparation shall print on the outer label of every package thereof instructions to store the preparation in a refrigerator at 35° to 50°F (2° to 10°C) and to avoid exposing it to freezing.
- SOR/82-769, s. 7
- SOR/97-12, s. 65
C.04.653 The packager/labeller of an insulin preparation that consists of a precipitate suspended in a buffered aqueous medium shall print on the inner label of every package thereof the statement “Shake Carefully”.
- SOR/82-769, s. 7
- SOR/97-12, s. 65
C.04.654 The packager/labeller of an insulin preparation may, in lieu of printing adequate directions for its use on both the inner and outer labels thereof as required by subparagraph C.04.019(a)(vii), print the descriptions for use in a descriptive circular prepared in accordance with section C.04.655, but in such case he shall
(a) enclose a copy of the circular in the package containing the preparation; and
(b) state on the outer label of the package that such a circular is enclosed therein.
- SOR/82-769, ss. 7, 9
- SOR/97-12, s. 65
C.04.655 The descriptive circular referred to in section C.04.654 shall include, at least, the following information:
(a) a statement that
(i) the treatment of diabetes mellitus requires medical supervision and review,
(ii) insulin preparations should be used only as determined by a physician for each patient in the light of blood-sugar and urinary-sugar findings, and
(iii) the physician’s instructions concerning diet, dosage, rest and exercise should be followed carefully;
(b) an outline of the procedure to be followed in withdrawing the insulin preparation from the vial, including techniques for sterilization of the syringe and needle, vial-stopper and site of injection;
(c) a statement explaining that injections should be subcutaneous, and not intravenous or intramuscular, and a caution against successive injections in any one site;
(d) a statement that doses are specified in terms of Units of potency per cubic centimetre and that the volume of each dose will depend upon the potency in terms of units per cubic centimetre stated on the label of the insulin preparation and that, for these reasons, it is important that the patient understand the markings on syringes;
(e) a brief explanation of hypoglycemia together with emergency measures suitable for use by patients and those caring for patients in the event of hypoglycemic reactions;
(f) a statement indicating the possibility of undesirable reactions associated with illness or infection, with the omission or loss of a meal, and with a shortage of the insulin preparation;
(g) a statement warning against using any other type of insulin preparation than that prescribed by the physician;
(h) a statement that the use of a package should not be commenced after the expiration date printed on the package;
(i) a statement that the contents should be used as continuously as practicable and that any vial from which a part of the contents has been withdrawn should be discarded in the event of its being in disuse for several weeks’ time;
(j) a statement stressing the importance of visiting a physician regularly and of carefully following his instructions;
(k) in the case of insulin preparations consisting of a clear, colourless or almost colourless solution, free from turbidity and from insoluble matter, a statement that if the contents of the vial become cloudy or turbid, use of that vial should be discontinued;
(l) in the case of insulin preparations consisting of a precipitate suspended in a buffered aqueous medium, a statement explaining that it is necessary to shake the vial carefully before withdrawing a dose, noting that if the contents have become lumpy or granular in appearance or have formed a deposit of particles on the wall of the container, the use of that vial should be discontinued;
(m) instructions that the insulin preparation should be stored in a refrigerator at 35° to 50°F (2° to 10°C) and should not be exposed to freezing; and
(n) in the case of Sulphated Insulin, a statement explaining that this insulin preparation is not for ordinary use, but is a chemically modified insulin which may be more effective than the usual insulin preparations in certain insulin-resistant or insulin-allergic diabetic patients.
- SOR/82-769, ss. 7, 10
C.04.656 (1) Notwithstanding section C.04.554, a person who holds an establishment licence may sell Insulin Injection made from zinc-insulin crystals contained in vials of approximately 20 cubic centimetre capacity each of which vials
(a) contains an excess volume sufficient to permit withdrawal of 20 cubic centimetres, and
(b) provides 500 International Units of insulin per cubic centimetre,
if
(c) notwithstanding section C.04.651, both the inner and outer labels are printed in black ink on white stock and overprinted in narrow brown and white diagonal stripes, of which there shall be at least five but not more than 20 to each inch;
(d) both the inner and the outer labels carry the statement “Warning — High Potency — Not for Ordinary Use”; and
(e) each package contains a descriptive circular that conforms to the requirements of section C.04.655 and, in addition, includes,
(i) at the beginning of the circular the statement:
“Warning — This insulin preparation contains 500 International Units of insulin in each cubic centimetre. Extreme caution must be observed in the measurement of doses because inadvertent overdose may result in irreversible shock. Serious consequences may result if it is used other than under constant medical supervision. Unless specifically prescribed it should never be used by patients to replace use of any other insulin preparations.”,
(ii) a statement that Insulin made from Zinc-Insulin crystals 500 International Units per cubic centimetre should not be administered intravenously, and
(iii) a statement giving information for the safe and effective use by physicians of the drug in insulin shock therapy and in the treatment of diabetic patients with high insulin resistance (daily requirement more than 200 International Units of insulin).
(2) [Repealed, SOR/95-203, s. 10]
- SOR/82-769, ss. 7, 11
- SOR/95-203, s. 10
- SOR/97-12, s. 64
Anterior Pituitary Extracts
- SOR/82-769, s. 14
C.04.675 Anterior pituitary extract shall include all natural products, prepared from the anterior lobe of the pituitary gland of animals, having physiological properties associated with the hormones of the anterior pituitary gland and their proper names shall be
(a) Adrenocorticotrophic Hormone, Corticotrophin,
(b) Thyrotrophic Hormone, Thyrotrophin,
(c) Growth Hormone Pituitary, Somatotrophin,
(d) Lactogenic Hormone, Prolactin,
(e) Gonadotrophic Hormone, Gonadotrophin, followed by qualifying words to indicate the gonadotrophic activity associated with the extract,
and if unpurified anterior pituitary extract
(f) Pituitary Extract Anterior Lobe followed by qualifying words to indicate the physiological properties associated with it.
- SOR/82-769, s. 13
C.04.676 Reference standards for anterior pituitary extract shall be
(a) the International Standard,
(b) where no International Standard exists, the Canadian Reference Standard shall be that established and kept by the Minister from whom portions for comparative testing may be had upon application, and
(c) where neither an International Standard nor a Canadian Reference Standard exists, a provisional reference standard that shall be a suitable quantity of the product submitted by the distributor referred to in paragraph C.01A.003(b) to the Minister for checking the uniformity of the product.
- SOR/82-769, s. 13
- SOR/97-12, s. 58
- SOR/2018-69, s. 27
C.04.677 Both the inner and outer labels of an anterior pituitary extract shall carry a statement of the potency in terms of the reference standard for anterior pituitary extract provided in section C.04.676 as determined by an acceptable method, except that where no reference standard for an anterior pituitary extract exists, the distributor referred to in paragraph C.01A.003(b) shall include, with every package of the anterior pituitary extract, an acceptable statement of the unit of potency and the method of assay used.
- SOR/82-769, s. 13
- SOR/97-12, s. 58
- SOR/97-543, s. 6
C.04.678 No person who holds an establishment licence shall sell corticotrophic hormones for subcutaneous or intramuscular use unless the preparation has been assayed by an acceptable method involving subcutaneous injection and, where the preparation is recommended for intravenous use, the label carries specific dosage instructions for that use.
- SOR/82-769, s. 13
- SOR/97-12, s. 64
C.04.679 No person shall sell as such adrenocorticotrophic hormone, thyrotrophic hormone, growth hormone pituitary, lactogenic hormone, or gonadotrophic hormone that is not acceptable free from any anterior pituitary extract other than the one for which it is named.
- SOR/82-769, s. 13
C.04.680 The outer label of a mixture of two or more of adrenocorticotrophic hormone, thyrotrophic hormone, growth hormone pituitary, lactogenic hormone and gonadotrophic hormone, or a mixture of any of those with pituitary extract anterior lobe, shall carry a declaration of the proper name and the amount of each component of the mixture.
- SOR/82-769, s. 13
- SOR/93-202, s. 22
C.04.681 The outer label of an anterior pituitary extract or mixture of anterior pituitary extracts shall carry a statement
(a) showing the species of animal from which the glands used in the preparation of the anterior pituitary extract were obtained,
(b) that it shall be stored at refrigerator temperature, and
(c) that, except in the case of gonadotrophic hormones, it is to be used only on the advice or on the prescription of a physician.
- SOR/82-769, s. 13
C.04.682 Both the inner and outer labels of adrenocorticotrophic hormone shall carry a statement indicating the route of administration, in addition to meeting the requirements of paragraphs C.04.681(a) and (b).
- SOR/82-769, ss. 13, 15
C.04.683 The expiration date for an anterior pituitary extract or mixture of anterior pituitary extracts shall be not more than two years after the date of passing a potency test.
- SOR/82-769, s. 13
DIVISION 5Drugs for Clinical Trials Involving Human Subjects
Interpretation
C.05.001 The definitions in this section apply in this Division.
- adverse drug reaction
adverse drug reaction means any noxious and unintended response to a drug that is caused by the administration of any dose of the drug. (réaction indésirable à une drogue)
- adverse event
adverse event means any adverse occurrence in the health of a clinical trial subject who is administered a drug, that may or may not be caused by the administration of the drug, and includes an adverse drug reaction. (incident thérapeutique)
- clinical trial
clinical trial means an investigation in respect of a drug for use in humans that involves human subjects and that is intended to discover or verify the clinical, pharmacological or pharmacodynamic effects of the drug, identify any adverse events in respect of the drug, study the absorption, distribution, metabolism and excretion of the drug, or ascertain the safety or efficacy of the drug. (essai clinique)
- drug
drug means a drug for human use that is to be tested in a clinical trial. (drogue)
- good clinical practices
good clinical practices means generally accepted clinical practices that are designed to ensure the protection of the rights, safety and well-being of clinical trial subjects and other persons, and the good clinical practices referred to in section C.05.010. (bonnes pratiques cliniques)
- import
import means to import a drug into Canada for the purpose of sale in a clinical trial. (importer)
- investigator’s brochure
investigator’s brochure means, in respect of a drug, a document containing the preclinical and clinical data on the drug that are described in paragraph C.05.005(e). (brochure du chercheur)
- protocol
protocol means a document that describes the objectives, design, methodology, statistical considerations and organization of a clinical trial. (protocole)
- qualified investigator
qualified investigator means the person responsible to the sponsor for the conduct of the clinical trial at a clinical trial site, who is entitled to provide health care under the laws of the province where that clinical trial site is located, and who is
(a) in the case of a clinical trial respecting a drug to be used for dental purposes only, a physician or dentist and a member in good standing of a professional medical or dental association; and
(b) in any other case, a physician and a member in good standing of a professional medical association. (chercheur qualifié)
- research ethics board
research ethics board means a body that is not affiliated with the sponsor, and
(a) the principal mandate of which is to approve the initiation of, and conduct periodic reviews of, biomedical research involving human subjects in order to ensure the protection of their rights, safety and well-being; and
(b) that has at least five members, that has a majority of members who are Canadian citizens or permanent residents under the Immigration and Refugee Protection Act, that is composed of both men and women and that includes at least
(i) two members whose primary experience and expertise are in a scientific discipline, who have broad experience in the methods and areas of research to be approved and one of whom is from a medical discipline or, if the clinical trial is in respect of a drug to be used for dental purposes only, is from a medical or dental discipline,
(ii) one member knowledgeable in ethics,
(iii) one member knowledgeable in Canadian laws relevant to the biomedical research to be approved,
(iv) one member whose primary experience and expertise are in a non-scientific discipline, and
(v) one member who is from the community or is a representative of an organization interested in the areas of research to be approved and who is not affiliated with the sponsor or the site where the clinical trial is to be conducted. (comité d’éthique de la recherche)
- serious adverse drug reaction
serious adverse drug reaction means an adverse drug reaction that requires in-patient hospitalization or prolongation of existing hospitalization, that causes congenital malformation, that results in persistent or significant disability or incapacity, that is life threatening or that results in death. (réaction indésirable grave à une drogue)
- serious unexpected adverse drug reaction
serious unexpected adverse drug reaction means a serious adverse drug reaction that is not identified in nature, severity or frequency in the risk information set out in the investigator’s brochure or on the label of the drug. (réaction indésirable grave et imprévue à une drogue)
- sponsor
sponsor means an individual, corporate body, institution or organization that conducts a clinical trial. (promoteur)
- SOR/2001-203, s. 4
- 2001, c. 27, s. 273
- SOR/2012-16, s. 1(F)
- SOR/2013-56, s. 1(F)
Application
C.05.002 (1) Subject to subsection (2), this Division applies to the sale or importation of drugs to be used for the purposes of clinical trials involving human subjects.
(2) Except for paragraph C.05.003(a), subsections C.05.006(2) and (3), paragraphs C.05.010(a) to (i), section C.05.011, subsections C.05.012(1) and (2), paragraphs C.05.012(3)(a) to (d) and (f) to (h), subsection C.05.012(4) and sections C.05.013, C.05.016 and C.05.017, this Division does not apply to the sale or importation of a drug for the purposes of a clinical trial authorized under subsection C.05.006(2).
- SOR/2001-203, s. 4
Prohibition
C.05.003 Despite sections C.01.014, C.08.002, C.08.002.02 and C.08.003, no person shall sell or import a drug for the purposes of a clinical trial unless
(a) the person is authorized under this Division;
(b) the person complies with this Division and sections C.01.015, C.01.036, C.01.037 to C.01.040, C.01.040.2, C.01.064 to C.01.067, C.01.070, C.01.131, C.01.133 to C.01.136, and C.01.435; and
(c) if the drug is to be imported, the person has a representative in Canada who is responsible for the sale of the drug.
- SOR/2001-203, s. 4
- SOR/2011-88, s. 7
General
C.05.004 Despite these Regulations, a sponsor may submit an application under this Division to sell or import a drug for the purposes of a clinical trial that contains a substance the sale of which is prohibited by these Regulations, if the sponsor establishes, on the basis of scientific information, that the inclusion of the substance in the drug may result in a therapeutic benefit for a human being.
- SOR/2001-203, s. 4
Application for Authorization
C.05.005 An application by a sponsor for authorization to sell or import a drug for the purposes of a clinical trial under this Division shall be submitted to the Minister, signed and dated by the sponsor’s senior medical or scientific officer in Canada and senior executive officer and shall contain the following information and documents:
(a) a copy of the protocol for the clinical trial;
(b) a copy of the statement, as it will be set out in each informed consent form, that states the risks and anticipated benefits arising to the health of clinical trial subjects as a result of their participation in the clinical trial;
(c) a clinical trial attestation, signed and dated by the sponsor’s senior medical or scientific officer in Canada and senior executive officer, containing
(i) the title of the protocol and the clinical trial number,
(ii) the brand name, the chemical name or the code for the drug,
(iii) the therapeutic and pharmacological classifications of the drug,
(iv) the medicinal ingredients of the drug,
(v) the non-medicinal ingredients of the drug,
(vi) the dosage form of the drug,
(vii) the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of the sponsor,
(viii) if the drug is to be imported, the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of the sponsor’s representative in Canada who is responsible for the sale of the drug,
(ix) for each clinical trial site, the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of the qualified investigator, if known at the time of submitting the application,
(x) for each clinical trial site, the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of the research ethics board that approved the protocol referred to in paragraph (a) and approved an informed consent form containing the statement referred to in paragraph (b), if known at the time of submitting the application, and
(xi) a statement
(A) that the clinical trial will be conducted in accordance with good clinical practices and these Regulations, and
(B) that all information contained in, or referenced by, the application is complete and accurate and is not false or misleading;
(d) the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of any research ethics board that has previously refused to approve the protocol referred to in paragraph (a), its reasons for doing so and the date on which the refusal was given, if known at the time of submitting the application;
(e) an investigator’s brochure that contains the following information, namely,
(i) the physical, chemical and pharmaceutical properties of the drug,
(ii) the pharmacological aspects of the drug, including its metabolites in all animal species tested,
(iii) the pharmacokinetics of the drug and the drug metabolism, including the biological transformation of the drug in all animal species tested,
(iv) any toxicological effects in any animal species tested under a single dose study, a repeated dose study or a special study in respect of the drug,
(v) any results of carcinogenicity studies in any animal species tested in respect of the drug,
(vi) any results of clinical pharmacokinetic studies of the drug,
(vii) any information regarding drug safety, pharmacodynamics, efficacy and dose responses of the drug that were obtained from previous clinical trials in humans, and
(viii) if the drug is a radiopharmaceutical as defined in section C.03.201, information regarding directions for preparing the radiopharmaceutical, the radiation dosimetry in respect of the prepared radiopharmaceutical and a statement of the storage requirements for the prepared radiopharmaceutical;
(f) if the drug contains a human-sourced excipient, including any used in the placebo,
(i) information that indicates the human-sourced excipient has been assigned a drug identification number under subsection C.01.014.2(1) or, in the case of a new drug, issued a notice of compliance under subsection C.08.004(1), as the case may be, or
(ii) in any other case, sufficient information to support the identity, purity, potency, stability and safety of the human-sourced excipient;
(g) if the drug has not been assigned a drug identification number under subsection C.01.014.2(1) or, in the case of a new drug, a notice of compliance has not been issued under section C.08.004 or C.08.004.01, the chemistry and manufacturing information in respect of the drug, including its site of manufacture; and
(h) the proposed date for the commencement of the clinical trial at each clinical trial site, if known at the time of submitting the application.
- SOR/2001-203, s. 4
- SOR/2011-88, s. 8
- SOR/2012-16, s. 2(F)
Authorization
C.05.006 (1) Subject to subsection (3), a sponsor may sell or import a drug, other than a drug described in subsection (2), for the purposes of a clinical trial if
(a) the sponsor has submitted to the Minister an application in accordance with section C.05.005;
(b) the Minister does not, within 30 days after the date of receipt of the application, send to the sponsor a notice in respect of the drug indicating that the sponsor may not sell or import the drug for any of the following reasons:
(i) that the information and documents in respect of the application
(A) were not provided in accordance with these Regulations, or
(B) are insufficient to enable the Minister to assess the safety and risks of the drug or the clinical trial, or
(ii) that based on an assessment of the application, an assessment of any information submitted under section C.05.009 or a review of any other information, the Minister has reasonable grounds to believe that
(A) the use of the drug for the purposes of the clinical trial endangers the health of a clinical trial subject or other person,
(B) the clinical trial is contrary to the best interests of a clinical trial subject, or
(C) the objectives of the clinical trial will not be achieved;
(c) for each clinical trial site, the sponsor has obtained the approval of the research ethics board in respect of the protocol referred to in paragraph C.05.005(a) and in respect of an informed consent form that contains the statement referred to in paragraph C.05.005(b); and
(d) before the sale or importation of the drug at a clinical trial site, the sponsor submits to the Minister the information referred to in subparagraphs C.05.005(c)(ix) and (x) and paragraphs C.05.005(d) and (h), if it was not submitted in respect of that clinical trial site at the time of submitting the application.
(2) Subject to subsection (3), a sponsor may sell or import a drug for the purposes of a clinical trial in respect of
(a) a new drug that has been issued a notice of compliance under subsection C.08.004(1), if the clinical trial is in respect of a purpose or condition of use for which the notice of compliance was issued; or
(b) a drug, other than a new drug, that has been assigned a drug identification number under subsection C.01.014.2(1), if the clinical trial is in respect of a use or purpose for which the drug identification number was assigned.
(3) A sponsor may not sell or import a drug for the purposes of a clinical trial
(a) during the period of any suspension made under section C.05.016 or C.05.017; or
(b) after a cancellation made under section C.05.016 or C.05.017.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 3(F)
Notification
C.05.007 If the sale or importation of a drug is authorized under this Division, the sponsor may make one or more of the following changes if the sponsor notifies the Minister in writing within 15 days after the date of the change:
(a) a change to the chemistry and manufacturing information that does not affect the quality or safety of the drug, other than a change for which an amendment is required by section C.05.008; and
(b) a change to the protocol that does not alter the risk to the health of a clinical trial subject, other than a change for which an amendment is required by section C.05.008.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 4(F)
Amendment
C.05.008 (1) Subject to subsections (4) and (5), when the sale or importation of a drug is authorized under this Division and the sponsor proposes to make an amendment referred to in subsection (2), the sponsor may sell or import the drug for the purposes of the clinical trial in accordance with the amended authorization, if the following conditions are met:
(a) the sponsor has submitted to the Minister an application for amendment in accordance with subsection (3);
(b) the Minister does not, within 30 days after the date of receipt of the application for amendment, send to the sponsor a notice in respect of the drug indicating that the sponsor may not sell or import the drug in accordance with the amendment for any of the following reasons, namely,
(i) that the information and documents in respect of the application for amendment
(A) were not provided in accordance with these Regulations, or
(B) are insufficient to enable the Minister to assess the safety and risks of the drug or the clinical trial, or
(ii) that based on an assessment of the application for amendment, an assessment of any information submitted under section C.05.009 or a review of any other information, the Minister has reasonable grounds to believe that
(A) the use of the drug for the purposes of the clinical trial endangers the health of a clinical trial subject or other person,
(B) the clinical trial is contrary to the best interests of a clinical trial subject, or
(C) the objectives of the clinical trial will not be achieved;
(c) before the sale or importation of the drug, the sponsor submits to the Minister
(i) for each clinical trial site, the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of the research ethics board that approved any amended protocol submitted under paragraph (3)(a) or approved any amended statement submitted under paragraph (3)(c), and
(ii) the name, address and telephone number and, if applicable, the facsimile number and electronic mail address of any research ethics board that has previously refused to approve any amendment to the protocol, its reasons for doing so and the date on which the refusal was given;
(d) before the sale or importation of the drug, the sponsor maintains records concerning
(i) the information referred to in paragraph C.05.005(h), and
(ii) the information referred to in subparagraph C.05.005(c)(ix), if any of that information has changed since it was submitted;
(e) before the sale or importation of the drug in accordance with the amended authorization, the sponsor ceases to sell or import the drug in accordance with the existing authorization; and
(f) the sponsor conducts the clinical trial in accordance with the amended authorization.
(2) For the purposes of subsection (1), amendments are
(a) amendments to the protocol that affect the selection, monitoring or dismissal of a clinical trial subject;
(b) amendments to the protocol that affect the evaluation of the clinical efficacy of the drug;
(c) amendments to the protocol that alter the risk to the health of a clinical trial subject;
(d) amendments to the protocol that affect the safety evaluation of the drug;
(e) amendments to the protocol that extend the duration of the clinical trial; and
(f) amendments to the chemistry and manufacturing information that may affect the safety or quality of the drug.
(3) The application for amendment referred to in subsection (1) shall contain a reference to the application submitted under section C.05.005 and shall contain the following documents and information:
(a) if the application is in respect of an amendment referred to in any of paragraphs (2)(a) to (e), a copy of the amended protocol that indicates the amendment, a copy of the protocol submitted under paragraph C.05.005(a), and the rationale for the amendment;
(b) if the application is in respect of an amendment referred to in paragraph (2)(e), a copy of the amended investigator’s brochure or an addendum to the investigator’s brochure that indicates the new information, including supporting toxicological studies and clinical trial safety data;
(c) if the application is in respect of an amendment referred to in any of paragraphs (2)(a) to (f) and, as a result of that amendment, it is necessary to amend the statement referred to in paragraph C.05.005(b), a copy of the amended statement that indicates the amendment; and
(d) if the application is in respect of an amendment referred to in paragraph (2)(f), a copy of the amended chemistry and manufacturing information that indicates the amendment, and the rationale for that amendment.
(4) If the sponsor is required to immediately make one or more of the amendments referred to in subsection (2) because the clinical trial or the use of the drug for the purposes of the clinical trial endangers the health of a clinical trial subject or other person, the sponsor may immediately make the amendment and shall provide the Minister with the information referred to in subsection (3) within 15 days after the date of the amendment.
(5) A sponsor may not sell or import a drug for the purposes of a clinical trial
(a) during the period of any suspension made under section C.05.016 or C.05.017; or
(b) after a cancellation made under section C.05.016 or C.05.017.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 5
Additional Information and Samples
C.05.009 If the information and documents submitted in respect of an application under section C.05.005 or an application for amendment under section C.05.008 are insufficient to enable the Minister to determine whether any of the reasons referred to in paragraph C.05.006(1)(b) or C.05.008(1)(b) exist, the Minister may require the sponsor to submit, within two days after receipt of the request, samples of the drug or additional information relevant to the drug or the clinical trial that are necessary to make the determination.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 6(F)
Sponsor’s Obligations
Good Clinical Practices
C.05.010 Every sponsor shall ensure that a clinical trial is conducted in accordance with good clinical practices and, without limiting the generality of the foregoing, shall ensure that
(a) the clinical trial is scientifically sound and clearly described in a protocol;
(b) the clinical trial is conducted, and the drug is used, in accordance with the protocol and this Division;
(c) systems and procedures that assure the quality of every aspect of the clinical trial are implemented;
(d) for each clinical trial site, the approval of a research ethics board is obtained before the clinical trial begins at the site;
(e) at each clinical trial site, there is no more than one qualified investigator;
(f) at each clinical trial site, medical care and medical decisions, in respect of the clinical trial, are under the supervision of the qualified investigator;
(g) each individual involved in the conduct of the clinical trial is qualified by education, training and experience to perform his or her respective tasks;
(h) written informed consent, given in accordance with the applicable laws governing consent, is obtained from every person before that person participates in the clinical trial but only after that person has been informed of
(i) the risks and anticipated benefits to his or her health arising from participation in the clinical trial, and
(ii) all other aspects of the clinical trial that are necessary for that person to make the decision to participate in the clinical trial;
(i) the requirements respecting information and records set out in section C.05.012 are met; and
(j) the drug is manufactured, handled and stored in accordance with the applicable good manufacturing practices referred to in Divisions 2 to 4 except sections C.02.019, C.02.025 and C.02.026.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 7(F)
Labelling
C.05.011 Despite any other provision of these Regulations respecting labelling, the sponsor shall ensure that the drug bears a label that sets out the following information in both official languages:
(a) a statement indicating that the drug is an investigational drug to be used only by a qualified investigator;
(b) the name, number or identifying mark of the drug;
(c) the expiration date of the drug;
(d) the recommended storage conditions for the drug;
(e) the lot number of the drug;
(f) the name and address of the sponsor;
(g) the protocol code or identification; and
(h) if the drug is a radiopharmaceutical as defined in section C.03.201, the information required by subparagraph C.03.202(1)(b)(vi).
- SOR/2001-203, s. 4
- SOR/2012-16, s. 8(F)
Records
C.05.012 (1) The sponsor shall record, handle and store all information in respect of a clinical trial in a way that allows its complete and accurate reporting as well as its interpretation and verification.
(2) The sponsor shall maintain complete and accurate records to establish that the clinical trial is conducted in accordance with good clinical practices and these Regulations.
(3) The sponsor shall maintain complete and accurate records in respect of the use of a drug in a clinical trial, including
(a) a copy of all versions of the investigator’s brochure for the drug;
(b) records respecting each change made to the investigator’s brochure, including the rationale for each change and documentation that supports each change;
(c) records respecting all adverse events in respect of the drug that have occurred inside or outside Canada, including information that specifies the indication for use and the dosage form of the drug at the time of the adverse event;
(d) records respecting the enrolment of clinical trial subjects, including information sufficient to enable all clinical trial subjects to be identified and contacted in the event that the sale of the drug may endanger the health of the clinical trial subjects or other persons;
(e) records respecting the shipment, receipt, disposition, return and destruction of the drug;
(f) for each clinical trial site, an undertaking from the qualified investigator that is signed and dated by the qualified investigator prior to the commencement of his or her responsibilities in respect of the clinical trial, that states that
(i) the qualified investigator will conduct the clinical trial in accordance with good clinical practices, and
(ii) the qualified investigator will immediately, on discontinuance of the clinical trial by the sponsor, in its entirety or at a clinical trial site, inform both the clinical trial subjects and the research ethics board of the discontinuance, provide them with the reasons for the discontinuance and advise them in writing of any potential risks to the health of clinical trial subjects or other persons;
(g) for each clinical trial site, a copy of the protocol, informed consent form and any amendment to the protocol or informed consent form that have been approved by the research ethics board for that clinical trial site; and
(h) for each clinical trial site, an attestation, signed and dated by the research ethics board for that clinical trial site, stating that it has reviewed and approved the protocol and informed consent form and that the board carries out its functions in a manner consistent with good clinical practices.
(4) The sponsor shall maintain all records referred to in this Division for a period of 15 years.
- SOR/2001-203, s. 4
- SOR/2022-18, s. 55
Submission of Information and Samples
C.05.013 (1) The Minister shall require a sponsor to submit, within two days after receipt of the request, information concerning the drug or the clinical trial, or samples of the drug, if the Minister has reasonable grounds to believe that
(a) the use of the drug for the purposes of the clinical trial endangers the health of a clinical trial subject or other person;
(b) the clinical trial is contrary to the best interests of a clinical trial subject;
(c) the objectives of the clinical trial will not be achieved;
(d) a qualified investigator is not respecting the undertaking referred to in paragraph C.05.012(3)(f); or
(e) information submitted in respect of the drug or the clinical trial is false or misleading.
(2) The Minister may require the sponsor to submit, within seven days after receipt of the request, any information or records kept under section C.05.012, or samples of the drug, in order to assess the safety of the drug or the health of clinical trial subjects or other persons.
- SOR/2001-203, s. 4
- SOR/2012-16, s. 9(F)
Serious Unexpected Adverse Drug Reaction Reporting
C.05.014 (1) During the course of a clinical trial, the sponsor shall inform the Minister of any serious unexpected adverse drug reaction in respect of the drug that has occurred inside or outside Canada as follows:
(a) if it is neither fatal nor life threatening, within 15 days after becoming aware of the information; and
(b) if it is fatal or life threatening, within seven days after becoming aware of the information.
(2) The sponsor shall, within eight days after having informed the Minister under paragraph (1)(b), submit to the Minister a complete report in respect of that information that includes an assessment of the importance and implication of any findings made.
(3) Sections C.01.016 to C.01.020 do not apply to drugs used for the purposes of a clinical trial.
- SOR/2001-203, s. 4
- SOR/2017-18, s. 16
Discontinuance of a Clinical Trial
C.05.015 (1) If a clinical trial is discontinued by the sponsor in its entirety or at a clinical trial site, the sponsor shall
(a) inform the Minister no later than 15 days after the date of the discontinuance;
(b) provide the Minister with the reason for the discontinuance and its impact on the proposed or ongoing clinical trials in respect of the drug conducted in Canada by the sponsor;
(c) as soon as possible, inform all qualified investigators of the discontinuance and of the reasons for the discontinuance, and advise them in writing of any potential risks to the health of clinical trial subjects or other persons; and
(d) in respect of each discontinued clinical trial site, stop the sale or importation of the drug as of the date of the discontinuance and take all reasonable measures to ensure the recovery of all unused quantities of the drug that have been sold.
(2) If the sponsor has discontinued the clinical trial in its entirety or at a clinical trial site, the sponsor may resume selling or importing the drug for the purposes of a clinical trial in its entirety or at a clinical trial site if, in respect of each clinical trial site where the sale or importation is to be resumed, the sponsor submits to the Minister the information referred to in subparagraphs C.05.005(c)(ix) and (x) and paragraphs C.05.005(d) and (h).
- SOR/2001-203, s. 4
Suspension and Cancellation
C.05.016 (1) Subject to subsection (2), the Minister shall suspend the authorization to sell or import a drug for the purposes of a clinical trial, in its entirety or at a clinical trial site, if the Minister has reasonable grounds to believe that
(a) the sponsor has contravened these Regulations or any provisions of the Act relating to the drug;
(b) any information submitted in respect of the drug or clinical trial is false or misleading;
(c) the sponsor has failed to comply with good clinical practices; or
(d) the sponsor has failed to provide
(i) information or samples of the drug as required under section C.05.009 or C.05.013, or
(ii) information or a report under section C.05.014.
(2) Subject to section C.05.017, the Minister shall not suspend an authorization referred to in subsection (1) unless
(a) the Minister has sent to the sponsor a written notice of the intention to suspend the authorization that indicates whether the authorization is to be suspended in its entirety or at a clinical trial site and the reason for the intended suspension;
(b) the sponsor has not, within 30 days after receipt of the notice referred to in paragraph (a), provided the Minister with information or documents that demonstrate that the authorization should not be suspended on the grounds that
(i) the situation giving rise to the intended suspension did not exist, or
(ii) the situation giving rise to the intended suspension has been corrected; and
(c) the Minister has provided the sponsor with the opportunity to be heard in paragraph (b).
(3) The Minister shall suspend the authorization by sending to the sponsor a written notice of suspension of the authorization that indicates the effective date of the suspension, whether the authorization is suspended in its entirety or at a clinical trial site and the reason for the suspension.
(4) If the Minister has suspended an authorization under subsection (1), the Minister shall
(a) reinstate the authorization in its entirety or at a clinical trial site, as the case may be, if within 30 days after the effective date of the suspension the sponsor provides the Minister with information or documents that demonstrate that the situation giving rise to the suspension has been corrected; or
(b) cancel the authorization in its entirety or at a clinical trial site, as the case may be, if within 30 days after the effective date of the suspension the sponsor has not provided the Minister with the information or documents referred to in paragraph (a).
- SOR/2001-203, s. 4
- SOR/2012-16, s. 10
C.05.017 (1) The Minister shall suspend an authorization to sell or import a drug for the purposes of a clinical trial, in its entirety or at a clinical trial site, before giving the sponsor an opportunity to be heard if the Minister has reasonable grounds to believe that it is necessary to do so to prevent injury to the health of a clinical trial subject or other person.
(2) The Minister shall suspend the authorization by sending to the sponsor a written notice of suspension of the authorization that indicates the effective date of the suspension, whether the authorization is suspended in its entirety or at a clinical trial site and the reason for the suspension.
(3) If the Minister has suspended an authorization under subsection (1), the Minister shall
(a) reinstate the authorization in its entirety or at a clinical trial site, as the case may be, if within 60 days after the effective date of the suspension the sponsor provides the Minister with information or documents that demonstrate that the situation giving rise to the suspension did not exist or that it has been corrected; or
(b) cancel the authorization in its entirety or at a clinical trial site, as the case may be, if within 60 days after the effective date of the suspension the sponsor has not provided the Minister with the information or documents referred to in paragraph (a).
- SOR/2001-203, s. 4
- SOR/2012-16, s. 11
DIVISION 6
Canadian Standard Drugs
- Conjugated Estrogens
- Conjugated Estrogens for Injection
- Conjugated Estrogens Tablets
- Digitoxin
- Digitoxin Tablets
- Digoxin
- Digoxin Elixir
- Digoxin Injection
- Digoxin Tablets
- Esterified Estrogens
- Esterified Estrogens Tablets
- Gelatin
- Thyroid
- SOR/80-544, s. 11
General
C.06.001 In this Division,
(a) solubility and specific gravity shall be determined at 25°C;
(b) tests for identity, quantitative tests for arsenic, lead, copper, zinc, fluorine, and sulphur dioxide, and limit tests shall be made by the official methods; and
(c) determination of physical and chemical constants shall be carried out by acceptable methods.
Conjugated Estrogens
C.06.002 [S]. Conjugated estrogens shall be the drug conjugated estrogens described in The Pharmacopeia of the United States of America, XVIII (1970), except that
(a) the dilute assay preparation A, assay preparations A and B and equilin reagent described therein shall be prepared by official method DO-29, Conjugated Estrogens, October 15, 1981; and
(b) the identification test described therein shall be performed by official method DO-29, Conjugated Estrogens, October 15, 1981.
- SOR/82-429, s. 5
Conjugated Estrogens for Injection
C.06.003 [S]. Conjugated estrogens for injection shall be the drug conjugated estrogens for injection described in The Pharmacopeia of the United States of America, XVIII (1970), except that
(a) the dilute assay preparation A, assay preparations A and B and equilin reagent described therein shall be prepared by official method DO-29, Conjugated Estrogens, October 15, 1981; and
(b) the identification test described therein shall be performed by official method DO-29, Conjugated Estrogens, October 15, 1981.
- SOR/82-429, s. 6
Conjugated Estrogens Tablets
C.06.004 [S]. Conjugated estrogens tablets shall be the drug conjugated estrogens tablets described in The Pharmacopeia of the United States of America, XVIII (1970), except that
(a) the dilute assay preparation A, assay preparations A and B and equilin reagent described therein shall be prepared by official method DO-29, Conjugated Estrogens, October 15, 1981; and
(b) the identification test described therein shall be performed by official method DO-29, Conjugated Estrogens, October 15, 1981.
- SOR/82-429, s. 7
C.06.100 and C.06.101 [Repealed, SOR/80-544, s. 12]
Digitoxin
C.06.120 [S]. Digitoxin shall be the drug digitoxin described in the Pharmacopeia of the United States of America.
Digitoxin Tablets
C.06.121 [S]. Digitoxin tablets shall be the drug digitoxin tablets described in the Pharmacopeia of the United States of America.
Digoxin
C.06.130 [S]. Digoxin shall be the drug digoxin described in the Pharmacopeia of the United States of America.
Digoxin Elixir
C.06.131 [S]. Digoxin Elixir shall be the drug digoxin elixir described in the Pharmacopeia of the United States of America.
Digoxin Injection
C.06.132 [S]. Digoxin injection shall be the drug digoxin injection described in the Pharmacopeia of the United States of America.
Digoxin Tablets
C.06.133 [S]. Digoxin tablets shall be the drug digoxin tablets described in the Pharmacopeia of the United States of America.
C.06.140 to C.06.142 [Repealed, SOR/80-544, s. 12]
C.06.150 to C.06.153 [Repealed, SOR/80-544, s. 12]
C.06.154 to C.06.156 [Repealed, SOR/80-544, s. 12]
C.06.157 to C.06.160 [Repealed, SOR/80-544, s. 12]
Esterified Estrogens
C.06.161 [S]. Esterified estrogens shall be the drug esterified estrogens described in the Pharmacopeia of the United States of America.
Esterified Estrogens Tablets
C.06.162 [S]. Esterified estrogens tablets shall be the drug esterified estrogens tablets described in the Pharmacopeia of the United States of America.
Gelatin
C.06.170 Gelatin shall be the drug gelatin described in the Pharmacopeia of the United States or the British Pharmacopeia.
C.06.180 to C.06.183 [Repealed, SOR/80-544, s. 12]
C.06.230 to C.06.233 [Repealed, SOR/80-544, s. 12]
C.06.240 to C.06.242 [Repealed, SOR/80-544, s. 12]
Thyroid
C.06.250 Thyroid shall be the cleaned, dried, powdered thyroid glands of domestic animals used for food, and shall contain not less than 0.17 per cent, and not more than 0.23 per cent iodine and no added iodine in either inorganic or organic form, and
(a) its characters are
Description, —
(i) General, — thyroid occurs as a cream-coloured, amorphous powder; the odour and taste are faint and meat-like, and
(ii) Microscopical, — when suitably mounted and examined under the microscope, thyroid shows the following: numerous smooth to striated hyaline fragments of colloids, of angular to irregular shape, that are colourless to pale yellow in water mounts, brown in Mallory’s stain and pink in solution of eosin, some of these fragments containing granules, minute vacuoles, crystalloidal bodies and cells; numerous irregular fragments of follicular epithelium staining brown with Mallory’s stain, the individual cells more or less polygonal to rounded-angular or irregularly cuboidal, often with prominent nuclei staining dark blue, their cytoplasm purplish with Delafield’s solution of haematoxylin; slender glistening segments of capillaries of closely undulate outline; numerous slender segments of neuraxons; numerous aggregates of particles of intercellular substance and slender, mostly straight connective tissue fibres staining blue to greenish blue with a mixture of Mallory’s stain and solution of phosphotungstic acid, the bundles of fibres often appearing reddish in Mallory’s stain; few glistening fragments of blood vessels with serrated or crenated ends as viewed in water mounts; and
(b) the tests for its purity are
(i) Inorganic iodine, — add to one gram of thyroid 10 millilitres of a saturated solution of zinc sulphate in water, shake, allow to stand five minutes, and filter through a fritted glass filter; add to five millilitres of the filtrate 0.5 millilitre of mucilage of starch and four drops each of a 10 per cent w/v solution of sodium nitrite in water and dilute sulphuric acid, shaking after each addition: no blue colour is produced, and
(ii) Moisture, — thyroid loses not more than six per cent moisture.
C.06.251 Thyroid shall be
(a) assayed by official method DO-26, Thyroid, October 15, 1981; and
(b) stored in a cool place and in a tightly-closed container.
- SOR/82-429, s. 8
C.06.252 [Repealed, SOR/80-544, s. 12]
C.06.260 to C.06.264 [Repealed, SOR/80-544, s. 12]
C.06.270 to C.06.280 [Repealed, SOR/80-544, s. 12]
DIVISION 7Sale of Drugs for the Purposes of Implementing the General Council Decision
Interpretation
C.07.001 The definitions in this section apply in this Division.
- Commissioner of Patents
Commissioner of Patents means the Commissioner of Patents appointed under subsection 4(1) of the Patent Act. (commissaire aux brevets)
- General Council Decision
General Council Decision has the meaning assigned by subsection 30(6) of the Act. (décision du Conseil général)
- SOR/2005-141, s. 1
Application
C.07.002 This Division applies to the sale of drugs for the purposes of implementing the General Council Decision.
- SOR/2005-141, s. 1
Application for Authorization
C.07.003 An application by a manufacturer for authorization to sell a drug under this Division shall be submitted to the Minister and shall contain the following information and documents:
(a) a statement that the manufacturer intends to file an application with the Commissioner of Patents under section 21.04 of the Patent Act;
(b) in respect of a new drug, the submission number and date of filing of the new drug submission or abbreviated new drug submission filed under section C.08.002 or C.08.002.1, respectively, and of any supplement filed under section C.08.003 in respect of the drug;
(c) in respect of a drug that is not a new drug,
(i) the application number and date of filing of the application that has been filed under section C.01.014.1 in respect of the drug, or
(ii) the drug identification number, if one has been assigned in respect of the drug pursuant to section C.01.014.2;
(d) for a drug in a solid dosage form, the manner in which the drug is marked in accordance with paragraph C.07.008(a) and evidence that such manner does not alter the safety and efficacy of the drug;
(e) for a drug in a dosage form that is not solid, the manner in which the immediate container is marked in accordance with paragraph C.07.008(a); and
(f) a sample of the label for the drug that includes the information required by paragraph C.07.008(c).
- SOR/2005-141, s. 1
Authorization
C.07.004 The Minister shall notify the manufacturer and the Commissioner of Patents for the purposes of paragraph 21.04(3)(b) of the Patent Act that the manufacturer’s drug meets the requirements of the Act and these Regulations if
(a) the manufacturer has submitted to the Minister an application in accordance with section C.07.003 and a copy of the application filed by the manufacturer with the Commissioner of Patents under section 21.04 of the Patent Act;
(b) in respect of a new drug, an examination of the new drug submission or abbreviated new drug submission or supplement to either submission by the Minister demonstrates that the submission or supplement complies with section C.08.002, C.08.002.1 or C.08.003, as the case may be, and section C.08.005.1;
(c) in respect of a drug that is not a new drug, a drug identification number has been assigned pursuant to section C.01.014.2; and
(d) the Minister is satisfied that the manufacturer and the drug comply with the Act and these Regulations.
- SOR/2005-141, s. 1
C.07.005 Despite sections C.01.014, C.08.002 and C.08.003, a manufacturer may sell a drug under this Division if
(a) the Minister has notified the Commissioner of Patents for the purposes of paragraph 21.04(3)(b) of the Patent Act that the drug meets the requirements of the Act and these Regulations; and
(b) the manufacturer has received authorization under section 21.04 of the Patent Act.
- SOR/2005-141, s. 1
C.07.006 Sections C.01.005 and C.01.014.1 to C.01.014.4 do not apply to new drugs sold under this Division.
- SOR/2005-141, s. 1
Notice to Commissioner of Patents
C.07.007 The Minister shall notify the manufacturer and the Commissioner of Patents for the purposes of paragraph 21.13(b) of the Patent Act in the event that the Minister is of the opinion that the manufacturer’s drug authorized to be sold under this Division has ceased to meet the requirements of the Act and these Regulations.
- SOR/2005-141, s. 1
Marking and Labelling
C.07.008 No person shall sell a drug under this Division unless
(a) the drug itself permanently bears the mark “XCL”, in the case of a drug in a solid dosage form, or the immediate container permanently bears the mark “XCL”, in the case of a drug in a dosage form that is not solid;
(b) the colour of the drug itself is significantly different from the colour of the version of the drug sold in Canada, in the case of a drug in a solid dosage form; and
(c) the label of the drug permanently bears the mark “XCL”, followed by the export tracking number assigned by the Minister under section C.07.009 and the words “FOR EXPORT UNDER THE GENERAL COUNCIL DECISION. NOT FOR SALE IN CANADA.” or “POUR EXPORTATION AUX TERMES DE LA DÉCISION DU CONSEIL GÉNÉRAL. VENTE INTERDITE AU CANADA.”
- SOR/2005-141, s. 1
C.07.009 The Minister shall assign an export tracking number to each drug in respect of which the Minister has notified the Commissioner of Patents under section C.07.004.
- SOR/2005-141, s. 1
Records
C.07.010 The manufacturer shall, with respect to a drug authorized to be sold under this Division,
(a) establish and maintain records, in a manner that enables an audit to be made, respecting the information referred to in subsection C.08.007(1), and retain those records for at least seven years from the day on which they were established; and
(b) provide to the Minister the summaries referred to in section C.08.008.
- SOR/2005-141, s. 1
- SOR/2014-125, s. 1
Notice to Minister
C.07.011 The manufacturer shall notify the Minister in writing not less than 15 days before commencing the manufacture of the first lot of a drug authorized to be sold under this Division and not less than 15 days before the exportation of each subsequent lot of the drug.
- SOR/2005-141, s. 1
DIVISION 8
New Drugs
C.08.001 For the purposes of the Act and this Division, new drug means a drug, other than a veterinary health product,
(a) that contains or consists of a substance, whether as an active or inactive ingredient, carrier, coating, excipient, menstruum or other component, that has not been sold as a drug in Canada for sufficient time and in sufficient quantity to establish in Canada the safety and effectiveness of that substance for use as a drug;
(b) that is a combination of two or more drugs, with or without other ingredients, and that has not been sold in that combination or in the proportion in which those drugs are combined in that drug, for sufficient time and in sufficient quantity to establish in Canada the safety and effectiveness of that combination and proportion for use as a drug; or
(c) with respect to which the manufacturer prescribes, recommends, proposes or claims a use as a drug, or a condition of use as a drug, including dosage, route of administration or duration of action, and that has not been sold for that use or condition of use in Canada for sufficient time and in sufficient quantity to establish in Canada the safety and effectiveness of that use or condition of use of that drug.
- SOR/95-172, s. 2
- SOR/2017-76, s. 10
C.08.001.1 For the purposes of this Division,
- Canadian reference product
Canadian reference product means
(a) a drug in respect of which a notice of compliance is issued under section C.08.004 or C.08.004.01 and which is marketed in Canada by the innovator of the drug,
(b) a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, where a drug in respect of which a notice of compliance has been issued under section C.08.004 or C.08.004.01 cannot be used for that purpose because it is no longer marketed in Canada, or
(c) a drug, acceptable to the Minister, that can be used for the purpose of demonstrating bioequivalence on the basis of pharmaceutical and, where applicable, bioavailability characteristics, in comparison to a drug referred to in paragraph (a); (produit de référence canadien)
- designated COVID-19 drug
designated COVID-19 drug means a new drug for which the purpose and conditions of use recommended by the manufacturer relate to COVID-19; (drogue désignée contre la COVID-19)
- pharmaceutical equivalent
pharmaceutical equivalent means a new drug that, in comparison with another drug, contains identical amounts of the identical medicinal ingredients, in comparable dosage forms, but that does not necessarily contain the same non-medicinal ingredients; (équivalent pharmaceutique)
- specifications
specifications means a detailed description of a new drug and of its ingredients and includes
(a) a statement of all properties and qualities of the ingredients that are relevant to the manufacture and use of the new drug, including the identity, potency and purity of the ingredients,
(b) a detailed description of the methods used for testing and examining the ingredients, and
(c) a statement of the tolerances associated with the properties and qualities of the ingredients. (spécifications)
- SOR/95-411, s. 3
- SOR/2011-88, s. 9
- SOR/2021-45, s. 13
C.08.002 (1) No person shall sell or advertise a new drug unless
(a) the manufacturer of the new drug has filed with the Minister a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission or an abbreviated extraordinary use new drug submission relating to the new drug that is satisfactory to the Minister;
(b) the Minister has issued, under section C.08.004 or C.08.004.01, a notice of compliance to the manufacturer of the new drug in respect of the submission; and
(c) the notice of compliance in respect of the submission has not been suspended under section C.08.006.
(d) [Repealed, SOR/2014-158, s. 10]
(2) A new drug submission shall contain sufficient information and material to enable the Minister to assess the safety and effectiveness of the new drug, including the following:
(a) a description of the new drug and a statement of its proper name or its common name if there is no proper name;
(b) a statement of the brand name of the new drug or the identifying name or code proposed for the new drug;
(c) a list of the ingredients of the new drug, stated quantitatively, and the specifications for each of those ingredients;
(d) a description of the plant and equipment to be used in the manufacture, preparation and packaging of the new drug;
(e) details of the method of manufacture and the controls to be used in the manufacture, preparation and packaging of the new drug;
(f) details of the tests to be applied to control the potency, purity, stability and safety of the new drug;
(g) detailed reports of the tests made to establish the safety of the new drug for the purpose and under the conditions of use recommended;
(h) substantial evidence of the clinical effectiveness of the new drug for the purpose and under the conditions of use recommended;
(i) a statement of the names and qualifications of all the investigators to whom the new drug has been sold;
(j) in the case of a new drug for veterinary use, a draft of every label to be used in connection with the new drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the new drug;
(j.1) in the case of a new drug for human use, mock-ups of every label to be used in connection with the new drug — including any package insert and any document that is provided on request and that sets out supplementary information on the use of the new drug — and mock-ups of the new drug’s packages;
(k) a statement of all the representations to be made for the promotion of the new drug respecting
(i) the recommended route of administration of the new drug,
(ii) the proposed dosage of the new drug,
(iii) the claims to be made for the new drug, and
(iv) the contra-indications and side effects of the new drug;
(l) a description of the dosage form in which it is proposed that the new drug be sold;
(m) evidence that all test batches of the new drug used in any studies conducted in connection with the submission were manufactured and controlled in a manner that is representative of market production;
(n) in the case of a new drug intended for administration to food-producing animals, the withdrawal period of the new drug; and
(o) in the case of a new drug for human use other than a designated COVID-19 drug, an assessment as to whether there is a likelihood that the new drug will be mistaken for another drug for which a drug identification number has been assigned due to a resemblance between the brand name that is proposed to be used in respect of the new drug and the brand name, common name or proper name of the other drug.
(2.1) A manufacturer may file, for a designated COVID-19 drug, a new drug submission that does not meet the requirements set out in paragraphs (2)(g) and (h) if the submission contains
(a) a statement that the submission contains evidence to establish that the requirement set out in paragraph (b) is met; and
(b) sufficient evidence to support the conclusion that the benefits associated with the designated COVID-19 drug outweigh the risks for the purpose and under the conditions of use recommended, with consideration given to the uncertainties relating to those benefits and risks as well as the public health need related to COVID-19.
(2.2) A manufacturer may file, for a designated COVID-19 drug for human use, a new drug submission that does not meet the requirements set out in paragraph (2)(j.1) if the submission contains a draft of every label to be used in connection with the designated COVID-19 drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the designated COVID-19 drug.
(2.3) If, at the time a new drug submission is filed for a designated COVID-19 drug, the manufacturer is unable to provide the Minister with information or material referred to in any of paragraphs (2)(e) to (k), (m) and (n) or in paragraph (2.1)(b) or subsection (2.2) or that information or material is incomplete, the manufacturer shall provide the Minister, at that time, with a plan that specifies how and when the manufacturer will provide the Minister with the missing information or material.
(2.4) Subsections (2.1) to (2.3) apply if
(a) the new drug submission contains a statement that the submission is for a designated COVID-19 drug; and
(b) the purpose and conditions of use specified in the new drug submission in respect of the designated COVID-19 drug relate only to COVID-19 and the submission contains a statement to that effect.
(2.5) Subsections (2.1) to (2.3) do not apply if the manufacturer is seeking a notice of compliance for a designated COVID-19 drug on the basis of a direct or indirect comparison between the designated COVID-19 drug and another designated COVID-19 drug.
(3) The manufacturer of a new drug shall, at the request of the Minister, provide the Minister, where for the purposes of a new drug submission the Minister considers it necessary to assess the safety and effectiveness of the new drug, with the following information and material:
(a) the names and addresses of the manufacturers of each of the ingredients of the new drug and the names and addresses of the manufacturers of the new drug in the dosage form in which it is proposed that the new drug be sold;
(b) samples of the ingredients of the new drug;
(c) samples of the new drug in the dosage form in which it is proposed that the new drug be sold; and
(d) any additional information or material respecting the safety and effectiveness of the new drug.
- SOR/85-143, s. 1
- SOR/93-202, s. 24
- SOR/95-411, s. 4
- SOR/2011-88, s. 10
- SOR/2014-158, s. 10
- SOR/2017-259, s. 22
- SOR/2018-69, s. 33(F)
- SOR/2018-84, s. 8(F)
- SOR/2021-45, s. 14
C.08.002.01 (1) A manufacturer of a new drug may file an extraordinary use new drug submission for the new drug if
(a) the new drug is intended for
(i) emergency use in situations where persons have been exposed to a chemical, biological, radiological or nuclear substance and action is required to treat, mitigate or prevent a life-threatening or other serious disease, disorder or abnormal physical state, or its symptoms, that results, or is likely to result, from that exposure, or
(ii) preventative use in persons who are at risk of exposure to a chemical, biological, radiological or nuclear substance that is potentially lethal or permanently disabling; and
(b) the requirements set out in paragraphs C.08.002(2)(g) and (h) cannot be met because
(i) exposing human volunteers to the substance referred to in paragraph (a) would be potentially lethal or permanently disabling, and
(ii) the circumstances in which exposure to the substance occurs are sporadic and infrequent.
(2) Subject to subsections (3) and (5), an extraordinary use new drug submission shall contain
(a) an attestation, signed and dated by the senior executive officer in Canada of the manufacturer filing the submission and by the manufacturer’s senior medical or scientific officer, certifying that the conditions referred to in paragraphs (1)(a) and (b) are met, together with sufficient supporting information to enable the Minister to determine that those conditions are met; and
(b) sufficient information and material to enable the Minister to assess the safety and effectiveness of the new drug, including the following:
(i) the information and material described in paragraphs C.08.002(2)(a) to (f), (i) to (m) and (o),
(ii) information respecting the pathophysiological mechanism for the toxicity of the chemical, biological, radiological or nuclear substance and describing the new drug’s ability to treat, mitigate or prevent that mechanism,
(iii) detailed reports of in vitro studies respecting the toxicity and activity of the new drug in relation to the recommended purpose,
(iv) detailed reports of studies, in an animal species that is expected to react with a response that is predictive for humans, establishing the safety of the new drug, and providing substantial evidence of its effect, when used for the purpose and under the conditions of use recommended,
(v) information confirming that the end point of animal studies is clearly related to the desired benefit in humans,
(vi) information demonstrating that there is a sufficient understanding of the pharmacokinetics and pharmacodynamics of the new drug in animals and in humans to enable inferences to be drawn in respect of humans so as to allow for the selection of an effective dose in humans,
(vii) information respecting the safety of the new drug in humans, including detailed reports of clinical trials, if any, establishing the safety of the new drug,
(viii) information, if any, respecting the effectiveness of the new drug in humans for the purpose or under the conditions of use recommended,
(ix) a plan for monitoring and establishing the safety and effectiveness of the new drug under the conditions of use recommended that includes procedures for gathering and analyzing data, and
(x) any available assessment reports regarding the new drug prepared by regulatory authorities in countries other than Canada.
(3) Reports referred to in subparagraph (2)(b)(iii) or information referred to in subparagraph (2)(b)(vi) may be omitted if the extraordinary use new drug submission includes a detailed scientific explanation as to why the reports are or the information is not available.
(4) Any information or material that is necessary to enable the Minister to assess the safety and effectiveness of the new drug shall, at the request of the Minister, be added to the extraordinary use new drug submission, including
(a) the names and addresses of the manufacturers of each of the ingredients of the new drug and the names and addresses of the manufacturers of the new drug in the dosage form in which it is proposed to be sold;
(b) samples of the ingredients of the new drug;
(c) samples of the new drug in the dosage form in which it is proposed to be sold; and
(d) any information omitted by virtue of subsection (3).
(5) If an extraordinary use new drug submission is in respect of a new purpose for a new drug for which a notice of compliance has been issued under section C.08.004, the information and material referred to in subparagraph (2)(b)(i) may be omitted unless any of it is different from that which was originally submitted.
- SOR/2011-88, s. 11
- SOR/2014-158, s. 11
C.08.002.02 Despite sections C.08.002 and C.08.003, no manufacturer or importer shall sell a new drug for extraordinary use in respect of which a notice of compliance has been issued under section C.08.004.01 except to
(a) the Government of Canada or the government of a province for the use of a department or agency of that government, on receipt of a written order signed by the minister responsible for the department or by the person in charge of the agency, or by their duly authorized representative; or
(b) a municipal government, or an institution of such a government, on receipt of a written order signed by a senior official of the government or institution or by his or her duly authorized representative.
- SOR/2011-88, s. 11
C.08.002.1 (1) A manufacturer of a new drug may file an abbreviated new drug submission or an abbreviated extraordinary use new drug submission for the new drug where, in comparison with a Canadian reference product,
(a) the new drug is the pharmaceutical equivalent of the Canadian reference product;
(b) the new drug is bioequivalent with the Canadian reference product, based on the pharmaceutical and, where the Minister considers it necessary, bioavailability characteristics;
(c) the route of administration of the new drug is the same as that of the Canadian reference product; and
(d) the conditions of use for the new drug fall within the conditions of use for the Canadian reference product.
(2) An abbreviated new drug submission or an abbreviated extraordinary use new drug submission shall contain sufficient information and material to enable the Minister to assess the safety and effectiveness of the new drug, including the following:
(a) the information and material described in
(i) paragraphs C.08.002(2)(a) to (f), (j) to (l) and (o), in the case of an abbreviated new drug submission, and
(ii) paragraphs C.08.002(2)(a) to (f), (j) to (l) and (o), and subparagraphs C.08.002.01(2)(b)(ix) and (x), in the case of an abbreviated extraordinary use new drug submission;
(b) information identifying the Canadian reference product used in any comparative studies conducted in connection with the submission;
(c) evidence from the comparative studies conducted in connection with the submission that the new drug is
(i) the pharmaceutical equivalent of the Canadian reference product, and
(ii) where the Minister considers it necessary on the basis of the pharmaceutical and, where applicable, bioavailability characteristics of the new drug, bioequivalent with the Canadian reference product as demonstrated using bioavailability studies, pharmacodynamic studies or clinical studies;
(d) evidence that all test batches of the new drug used in any studies conducted in connection with the submission were manufactured and controlled in a manner that is representative of market production; and
(e) for a drug intended for administration to food-producing animals, sufficient information to confirm that the withdrawal period is identical to that of the Canadian reference product.
(3) The manufacturer of a new drug shall, at the request of the Minister, provide the Minister, where for the purposes of an abbreviated new drug submission or an abbreviated extraordinary use new drug submission the Minister considers it necessary to assess the safety and effectiveness of the new drug, with the following information and material:
(a) the names and addresses of the manufacturers of each of the ingredients of the new drug and the names and addresses of the manufacturers of the new drug in the dosage form in which it is proposed that the new drug be sold;
(b) samples of the ingredients of the new drug;
(c) samples of the new drug in the dosage form in which it is proposed that the new drug be sold; and
(d) any additional information or material respecting the safety and effectiveness of the new drug.
(4) For the purposes of this section, in the case of an abbreviated new drug submission, a new drug for extraordinary use in respect of which a notice of compliance has been issued under section C.08.004.01 is not a Canadian reference product.
- SOR/95-411, s. 5
- SOR/2011-88, s. 12
- SOR/2014-158, s. 12
C.08.003 (1) Despite section C.08.002, no person shall sell a new drug in respect of which a notice of compliance has been issued to the manufacturer of that new drug and has not been suspended under section C.08.006, if any of the matters specified in subsection (2) are significantly different from the information or material contained in the new drug submission, extraordinary use new drug submission, abbreviated new drug submission or abbreviated extraordinary use new drug submission, unless
(a) the manufacturer of the new drug has filed with the Minister a supplement to that submission;
(b) the Minister has issued a notice of compliance to the manufacturer of the new drug in respect of the supplement; and
(c) the notice of compliance in respect of the supplement has not been suspended under section C.08.006.
(d) [Repealed, SOR/2014-158, s. 13]
(2) The matters specified for the purposes of subsection (1), in relation to the new drug, are the following:
(a) the description of the new drug;
(b) the brand name of the new drug or the identifying name or code proposed for the new drug;
(c) the specifications of the ingredients of the new drug;
(d) the plant and equipment used in manufacturing, preparation and packaging the new drug;
(e) the method of manufacture and the controls used in manufacturing, preparation and packaging the new drug;
(f) the tests applied to control the potency, purity, stability and safety of the new drug;
(g) the labels used in connection with the new drug;
(g.1) in the case of a new drug for human use, its packages;
(h) the representations made with regard to the new drug respecting
(i) the recommended route of administration of the new drug,
(ii) the dosage of the new drug,
(iii) the claims made for the new drug,
(iv) the contra-indications and side effects of the new drug, and
(v) the withdrawal period of the new drug; and
(i) the dosage form in which it is proposed that the new drug be sold.
(3) A supplement to a submission referred to in subsection (1), with respect to the matters that are significantly different from those contained in the submission, shall contain sufficient information and material to enable the Minister to assess the safety and effectiveness of the new drug in relation to those matters.
(3.1) A supplement to a submission referred to in subsection (1) shall contain, as the case may be,
(a) if, due to a matter specified in subsection (2) — other than the brand name of a new drug for human use — that the supplement concerns, it is necessary to modify a new drug’s labels:
(i) in the case of a new drug for veterinary use, a draft of every label to be used in connection with the new drug, including any package insert and any document that is provided on request and that sets out supplementary information on the use of the new drug, or
(ii) in the case of a new drug for human use, mock-ups of every label to be used in connection with the new drug — including any package insert and any document that is provided on request and that sets out supplementary information on the use of the new drug — and mock-ups of the new drug’s packages; or
(b) if the supplement concerns the brand name of a new drug for human use:
(i) an assessment as to whether there is a likelihood that the new drug will be mistaken for another drug for which a drug identification number has been assigned due to a resemblance between the brand name that is proposed to be used in respect of the new drug and the brand name, common name or proper name of the other drug, and
(ii) mock-ups of every label to be used in connection with the new drug — including any package insert and any document that is provided on request and that sets out supplementary information on the use of the new drug — and mock-ups of the new drug’s packages.
(4) If a supplement to an extraordinary use new drug submission or an abbreviated extraordinary use new drug submission concerns a matter specified in subparagraph (2)(h)(iii), the supplement shall contain the attestation and supporting information referred to in paragraph C.08.002.01(2)(a).
- SOR/85-143, s. 2
- SOR/93-202, s. 25
- SOR/95-411, s. 6
- SOR/2011-88, s. 13
- SOR/2014-158, s. 13
- SOR/2017-259, s. 23
- SOR/2018-69, s. 33(F)
- SOR/2018-84, s. 9(F)
C.08.003.01 (1) The following definitions apply in this section.
- submission
submission means
(a) a new drug submission that is filed under section C.08.002;
(b) an extraordinary use new drug submission that is filed under section C.08.002.01;
(c) an abbreviated new drug submission that is filed under section C.08.002.1; or
(d) an abbreviated extraordinary use new drug submission that is filed under section C.08.002.1. (présentation)
- supplement
supplement means a supplement to a submission that is filed under section C.08.003. (supplément)
(2) Despite sections C.08.002, C.08.002.01, C.08.002.1 and C.08.003 and subject to subsection (3), the manufacturer of a new drug is not permitted to file a submission or supplement for the new drug on the basis of a direct or indirect comparison to any new drug in respect of which an authorization was issued under the ISAD Interim Order.
(3) Subsection (2) does not prevent the manufacturer of a new drug from filing a submission or supplement for the new drug on the basis of a direct or indirect comparison to another new drug in respect of which an authorization was issued under the ISAD Interim Order if
(a) a notice of compliance is issued under section C.08.004 or C.08.004.01 in respect of a submission or supplement for that other new drug; and
(b) the comparison is in respect of the matters that were approved by the notice of compliance.
C.08.003.1 In examining a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission or a supplement to any of those submissions, the Minister may examine any information or material filed with the Minister by any person pursuant to Division 5 or section C.08.002, C.08.002.01, C.08.002.1, C.08.003, C.08.005 or C.08.005.1 to establish the safety and effectiveness of the new drug for which the submission or supplement has been filed.
- SOR/95-411, s. 6
- SOR/2001-203, s. 5
- SOR/2011-88, s. 14
C.08.004 (1) Subject to section C.08.004.1, the Minister shall, after completing an examination of a new drug submission or abbreviated new drug submission or a supplement to either submission,
(a) if that submission or supplement complies with section C.08.002, C.08.002.1 or C.08.003, as the case may be, and section C.08.005.1, issue a notice of compliance; or
(b) if that submission or supplement does not comply with section C.08.002, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, issue a notice to the manufacturer to that effect.
(2) If a new drug submission or an abbreviated new drug submission or a supplement to either submission does not comply with section C.08.002, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, the manufacturer who filed the submission or supplement may amend the submission or supplement by filing additional information or material within 90 days after the day on which the Minister issues a notice to the manufacturer under paragraph C.08.004(1)(b) or within any longer period specified by the Minister.
(3) Subject to section C.08.004.1, the Minister shall, after completing an examination of any additional information or material filed in respect of a new drug submission or an abbreviated new drug submission or a supplement to either submission,
(a) if that submission or supplement complies with section C.08.002, C.08.002.1 or C.08.003, as the case may be, and section C.08.005.1, issue a notice of compliance; or
(b) if that submission or supplement does not comply with section C.08.002, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, issue a notice to the manufacturer to that effect.
(4) A notice of compliance issued in respect of a new drug on the basis of information and material contained in a submission filed pursuant to section C.08.002.1 shall state the name of the Canadian reference product referred to in the submission and shall constitute a declaration of equivalence for that new drug.
- SOR/84-267, ss. 1 to 3
- SOR/85-143, s. 3
- SOR/86-1009, s. 1
- SOR/86-1101, s. 1
- SOR/88-42, s. 1
- SOR/88-257, s. 1
- SOR/95-411, s. 6
- SOR/2019-62, s. 1
C.08.004.01 (1) Subject to section C.08.004.1, the Minister shall, after completing an examination of an extraordinary use new drug submission or an abbreviated extraordinary use new drug submission or a supplement to either submission,
(a) if that submission or supplement complies with section C.08.002.01, C.08.002.1 or C.08.003, as the case may be, and section C.08.005.1, issue a notice of compliance; or
(b) if that submission or supplement does not comply with section C.08.002.01, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, issue a notice to the manufacturer to that effect.
(2) If an extraordinary use new drug submission or an abbreviated extraordinary use new drug submission or a supplement to either submission does not comply with section C.08.002.01, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, the manufacturer who filed the submission or supplement may amend the submission or supplement by filing additional information or material within 90 days after the day on which the Minister issues a notice to the manufacturer under paragraph C.08.004.01(1)(b) or within any longer period specified by the Minister.
(3) Subject to section C.08.004.1, the Minister shall, after completing an examination of any additional information or material filed in respect of an extraordinary use new drug submission or an abbreviated extraordinary use new drug submission or a supplement to either submission,
(a) if that submission or supplement complies with section C.08.002.01, C.08.002.1 or C.08.003, as the case may be, and section C.08.005.1, issue a notice of compliance; or
(b) if that submission or supplement does not comply with section C.08.002.01, C.08.002.1 or C.08.003, as the case may be, or section C.08.005.1, issue a notice to the manufacturer to that effect.
(4) A notice of compliance issued in respect of a new drug for extraordinary use on the basis of information and material contained in a submission filed pursuant to section C.08.002.1 shall state the name of the Canadian reference product referred to in the submission and shall constitute a declaration of equivalence for that new drug.
- SOR/2011-88, s. 15
- SOR/2019-62, s. 2
C.08.004.1 (1) The following definitions apply in this section.
- abbreviated new drug submission
abbreviated new drug submission includes an abbreviated extraordinary use new drug submission. (présentation abrégée de drogue nouvelle)
- innovative drug
innovative drug means a drug that contains a medicinal ingredient not previously approved in a drug by the Minister and that is not a variation of a previously approved medicinal ingredient such as a salt, ester, enantiomer, solvate or polymorph. (drogue innovante)
- new drug submission
new drug submission includes an extraordinary use new drug submission. (présentation de drogue nouvelle)
- pediatric populations
pediatric populations means the following groups: premature babies born before the 37th week of gestation; full-term babies from 0 to 27 days of age; and all children from 28 days to 2 years of age, 2 years plus 1 day to 11 years of age and 11 years plus 1 day to 18 years of age. (population pédiatrique)
(1.1) For the purposes of the definition innovative drug in subsection (1), a medicinal ingredient is not considered to be approved in a drug by reason of the Minister having issued or amended an authorization under the ISAD Interim Order in respect of a COVID-19 drug that contains the medicinal ingredient.
(2) The purpose of this section is to implement Articles 20.48 and 20.49 of the Agreement between Canada, the United States of America and the United Mexican States, as defined in the definition Agreement in section 2 of the Canada–United States–Mexico Agreement Implementation Act, and paragraph 3 of Article 39 of the Agreement on Trade-related Aspects of Intellectual Property Rights set out in Annex 1C to the Agreement Establishing the World Trade Organization, as defined in the definition Agreement in subsection 2(1) of the World Trade Organization Agreement Implementation Act.
(3) If a manufacturer seeks a notice of compliance for a new drug on the basis of a direct or indirect comparison between the new drug and an innovative drug,
(a) the manufacturer may not file a new drug submission, a supplement to a new drug submission, an abbreviated new drug submission or a supplement to an abbreviated new drug submission in respect of the new drug before the end of a period of six years after the day on which the first notice of compliance was issued to the innovator in respect of the innovative drug; and
(b) the Minister shall not approve that submission or supplement and shall not issue a notice of compliance in respect of the new drug before the end of a period of eight years after the day on which the first notice of compliance was issued to the innovator in respect of the innovative drug.
(4) The period specified in paragraph (3)(b) is lengthened to eight years and six months if
(a) the innovator provides the Minister with the description and results of clinical trials relating to the use of the innovative drug in relevant pediatric populations in its first new drug submission for the innovative drug or in any supplement to that submission that is filed within five years after the issuance of the first notice of compliance for that innovative drug; and
(b) before the end of a period of six years after the day on which the first notice of compliance was issued to the innovator in respect of the innovative drug, the Minister determines that the clinical trials were designed and conducted for the purpose of increasing knowledge of the use of the innovative drug in those pediatric populations and this knowledge would there-by provide a health benefit to members of those populations.
(5) Subsection (3) does not apply if the innovative drug is not being marketed in Canada.
(6) Paragraph (3)(a) does not apply to a manufacturer if the innovator consents to the filing of a new drug submission, a supplement to a new drug submission, an abbreviated new drug submission or a supplement to an abbreviated new drug submission by the manufacturer before the end of the period of six years specified in that paragraph.
(7) Paragraph (3)(a) does not apply to a manufacturer if the manufacturer files an application for authorization to sell its new drug under section C.07.003.
(8) Paragraph (3)(b) does not apply to a manufacturer if the innovator consents to the issuance of a notice of compliance to the manufacturer before the end of the period of eight years specified in that paragraph or of eight years and six months specified in subsection (4).
(9) The Minister shall maintain a register of innovative drugs that includes information relating to the matters specified in subsections (3) and (4).
- SOR/95-411, s. 6
- SOR/2006-241, s. 1
- SOR/2011-88, s. 16
- SOR/2014-124, s. 1
- SOR/2020-74, s. 6
- SOR/2021-45, s. 16
C.08.005 (1) Subject to subsection (1.1) and notwithstanding sections C.08.002 and C.08.003, a manufacturer of a new drug may sell it to a qualified investigator to be used solely for the purpose of clinical testing to obtain evidence with respect to the safety, dosage and effectiveness of that new drug, when the following conditions are met:
(a) before the sale, the manufacturer has filed with the Minister, in compliance with section C.08.005.1, a preclinical submission containing information and material respecting
(i) the brand name of the new drug or the identifying name or code proposed for the new drug,
(ii) the chemical structure or other specific identification of the composition of the new drug,
(iii) the source of the new drug,
(iv) a detailed protocol of the clinical testing,
(v) the results of investigations made to support the clinical use of the new drug,
(vi) the contra-indications and precautions known in respect of the new drug and the suggested treatment of overdosage of the new drug,
(vii) all ingredients of the new drug, stated quantitatively,
(viii) the methods, equipment, plant and controls used in the manufacture, processing and packaging of the new drug,
(ix) the tests applied to control the potency, purity and safety of the new drug, and
(x) the names and qualifications of all investigators to whom the drug is to be sold and the names of all institutions in which the clinical testing is to be carried out;
(b) the Minister has not, within 60 days after the date of receipt of the preclinical submission, sent by registered mail to the manufacturer a notice in respect of that new drug indicating that the preclinical submission is not satisfactory;
(c) all inner labels and outer labels used in conjunction with the sale of the new drug to qualified investigators carry the statements
(i) “Investigational Drug” or “Drogue de recherche”, and
(ii) “To Be Used By Qualified Investigators Only” or “Réservée uniquement à l’usage de chercheurs compétents”;
(d) before the sale, the manufacturer ascertains that every qualified investigator to whom the new drug is to be sold
(i) has the facilities for the clinical testing to be conducted by the investigator, and
(ii) has received the information and material referred to in subparagraphs (a)(i) to (vi); and
(e) every qualified investigator to whom the new drug is to be sold has agreed in writing with the manufacturer that the investigator will
(i) not use the new drug or permit it to be used other than for clinical testing,
(ii) not permit the new drug to be used by any person other than the investigator except under the investigator’s direction,
(iii) report immediately to that manufacturer and, if so required by the Minister, report to the Minister all serious adverse reactions encountered during the clinical testing, and
(iv) account to the manufacturer for all quantities of the new drug received, where so requested by the manufacturer.
(1.1) This section applies only in respect of a new drug for veterinary use.
(2) Notwithstanding subsection (1), no manufacturer shall sell a new drug to a qualified investigator unless that manufacturer has, in respect of all previous sales of that new drug to any qualified investigator,
(a) kept accurate records of the distribution of that new drug and of the results of the clinical testing and has made those records available to the Minister for inspection on the request of the Minister; and
(b) immediately reported to the Minister all information he has obtained with respect to serious adverse reactions.
(3) The Minister may notify the manufacturer of a new drug that sales of that new drug to qualified investigators are prohibited if, in the opinion of the Minister, it is in the interest of public health to do so.
(4) Notwithstanding subsection (1), no manufacturer shall sell a new drug to a qualified investigator if the Minister has notified the manufacturer of that drug that such sales are prohibited.
(5) Paragraph (1)(c) does not apply to a radiopharmaceutical as defined in section C.03.201 or to a component or kit as defined in section C.03.205.
- SOR/79-236, s. 5
- SOR/85-143, s. 4
- SOR/87-511, s. 1
- SOR/93-202, s. 26
- SOR/95-411, s. 7
- SOR/2001-203, s. 6
- SOR/2018-69, s. 27
C.08.005.1 (1) Every manufacturer who files a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission, a supplement to any of those submissions or a submission for the clinical testing of a new drug for veterinary use shall, in addition to any information and material that is required under section C.08.002, C.08.002.01, C.08.002.1, C.08.003 or C.08.005, include in the submission or supplement
(a) [Repealed, SOR/2018-84, s. 10]
(b) a sectional report in respect of each human, animal and in vitro study included in the submission or supplement;
(c) a comprehensive summary of each human, animal and in vitro study referred to or included in the submission or supplement; and
(d) a submission certificate in respect of all information and material contained in the submission or supplement and any additional information or material filed to amend the submission or supplement.
(2) A sectional report referred to in paragraph (1)(b) shall include
(a) a summary of each study included in the submission or supplement;
(b) a summary of any additional information or material filed to amend the submission or supplement; and
(c) where raw data is available to the manufacturer in respect of a study,
(i) a summary of the data,
(ii) a cross-referencing of the data to the relevant portions of the sectional report,
(iii) a description of the conditions under which the experiments from which the data were obtained were conducted,
(iv) the details of the data treatment process, and
(v) the results and conclusions of the study.
(3) The comprehensive summary referred to in paragraph (1)(c) shall include a summary of the methods used, results obtained and conclusions arrived at in respect of all studies referred to or included in the submission or supplement and shall be cross-referenced to the relevant portions of the sectional reports.
(4) The submission certificate referred to in paragraph (1)(d) shall
(a) certify that all information and material included in the submission or supplement and any additional information or material filed to amend the submission or supplement are accurate and complete, and that the sectional reports and the comprehensive summary correctly represent the information and material referred to or included in the submission or supplement; and
(b) be signed and dated by
(i) the senior executive officer in Canada of the manufacturer filing the submission or supplement, and
(ii) the senior medical or scientific officer of the manufacturer.
(5) [Repealed, SOR/2018-84, s. 10]
(6) Every manufacturer who has filed a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission, a supplement to any of those submissions or a submission for the clinical testing of a new drug for veterinary use and who has any relating clinical case reports or raw data that were not included in the submission or supplement shall keep those reports or data and shall, within 30 days after receiving a written request from the Minister, submit them to the Minister.
- SOR/85-143, s. 5
- SOR/92-543, s. 1
- SOR/94-689, s. 2(F)
- SOR/95-411, s. 8
- SOR/2001-203, s. 7
- SOR/2011-88, s. 17
- SOR/2018-84, s. 10
C.08.006 (1) For the purposes of subsection (2), evidence or new information obtained by the Minister includes any information or material filed by any person under Division 5 or section C.08.002, C.08.002.01, C.08.002.1, C.08.003, C.08.005 or C.08.005.1.
(2) The Minister may, by notice to a manufacturer, suspend, for a definite or indefinite period, a notice of compliance issued to that manufacturer in respect of a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission or a supplement to any of those submissions if the Minister considers
(a) that the drug is not safe for the use represented in the submission or supplement, as shown by evidence obtained from
(i) clinical or other experience not reported in the submission or supplement or not available to the Minister at the time the notice of compliance was issued, or
(ii) tests by new methods or tests by methods not reasonably applicable at the time the notice of compliance was issued;
(b) that, upon the basis of new information obtained after the issuance of the notice of compliance, there is lack of substantial evidence that the drug will have the effect it is represented to have under the conditions of use prescribed, recommended or proposed by the manufacturer;
(c) that the submission or supplement contained an untrue statement of material fact;
(d) that the manufacturer has failed to establish a system for maintaining required records or has repeatedly or deliberately failed to maintain such records;
(e) that, on the basis of new information obtained after the issuance of the notice of compliance, the methods, equipment, plant and controls used in the manufacturing, processing and packaging of the drug are inadequate to assure and preserve the identity, strength, quality or purity of the new drug;
(f) that, on the basis of new information obtained after the issuance of the notice of compliance, the labelling of the drug is false or misleading or incomplete in any particular and that this defect was not corrected by the manufacturer upon receipt of a written notice from the Minister specifying the respect in which the labelling is false or misleading or incomplete; or
(g) that, in the case of a new drug for extraordinary use, the manufacturer has not adhered to the plan referred to in subparagraph C.08.002.01(2)(b)(ix).
(3) The Minister may, by notice to a manufacturer, suspend for a definite or indefinite period a notice of compliance issued to that manufacturer in respect of a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission or a supplement to any of those submissions if, after the Minister has, under section 21.31 of the Act, ordered the holder of a therapeutic product authorization referred to in subparagraph C.01.052(1)(a)(iii) to conduct an assessment of the new drug in order to provide evidence establishing that the benefits associated with the drug outweigh the risks of injury to health,
(a) the holder fails to comply with the order; or
(b) the holder complies with the order but the Minister determines that the results of the assessment are not sufficient to establish that the benefits associated with the drug outweigh the risks of injury to health.
- SOR/95-411, s. 9
- SOR/2001-203, s. 8
- SOR/2011-88, s. 18
- SOR/2018-69, s. 27
- SOR/2018-84, s. 11
- SOR/2020-262, s. 5
C.08.007 (1) Where a manufacturer has received a notice of compliance issued in respect of a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission, an abbreviated extraordinary use new drug submission or a supplement to any of those submissions, the manufacturer shall establish and maintain records, in a manner that enables an audit to be made, respecting
(a) animal or clinical experience, studies, investigations and tests conducted by the manufacturer or reported to him by any person concerning that new drug;
(b) reports from the scientific literature or the bibliography therefrom that are available to him concerning that new drug;
(c) experience, investigations, studies and tests involving the chemical or physical properties or any other properties of that new drug;
(d) any substitution of another substance for that new drug or any mixing of another substance with that new drug;
(e) any error in the labelling of that new drug or in the use of the labels designed for that new drug;
(f) any bacteriological or any significant chemical or physical or other change or deterioration in any lot of that new drug;
(g) any failure of one or more distributed lots of the new drug to meet the specifications established for that new drug in the submission or supplement; and
(h) any unusual failure in efficacy of that new drug.
(i) [Repealed, SOR/95-521, s. 3]
(1.1) The manufacturer shall retain the records respecting the information referred to in subsection (1) for at least seven years from the day on which they were established.
(2) A manufacturer or importer who sells a new drug for extraordinary use in accordance with section C.08.002.02 shall retain the written order for at least 15 years from the day on which the order was filled.
- SOR/95-411, s. 10
- SOR/95-521, s. 3
- SOR/2011-88, s. 19
- SOR/2014-125, s. 2
C.08.008 No manufacturer shall sell a new drug unless the manufacturer has, with respect to all the manufacturer’s previous sales of that new drug, furnished to the Minister
(a) a summary of a record respecting any information referred to in paragraphs C.08.007(1)(a) to (c), on receipt of a request from the Minister for the summary;
(b) a summary of a record respecting any information referred to in paragraph C.08.007(1)(d) to (f), immediately on the manufacturer establishing the record; and
(c) a summary of a record respecting any information referred to in paragraph C.08.007(1)(g) or (h), within 15 days after the day on which the manufacturer established the record.
- SOR/95-411, s. 11
- SOR/95-521, s. 4
- SOR/2014-125, s. 3
- SOR/2018-84, s. 12
C.08.008.1 Where a manufacturer has received a notice of compliance issued in respect of an extraordinary use new drug submission, an abbreviated extraordinary use new drug submission or a supplement to either of those submissions, the manufacturer
(a) shall adhere to the plan referred to in subparagraph C.08.002.01(2)(b)(ix); and
(b) shall, before the first day of October in each year and whenever requested to do so by the Minister for the purposes of assessing the safety and effectiveness of the drug to which the notice of compliance relates, provide a report on the use of the drug, including a critical analysis of any available updated information respecting the drug’s safety and effectiveness.
- SOR/2011-88, s. 20
C.08.009 (1) Where the Minister has decided
(a) to notify the manufacturer of a new drug for veterinary use that the sale of that drug to qualified investigators is prohibited, or
(b) to suspend a notice of compliance issued under section C.08.004 or C.08.004.01,
the manufacturer, if dissatisfied with that decision, may require the Minister to provide him with the reasons for the decision.
(2) Where the manufacturer has received the reasons for a decision of the Minister pursuant to subsection (1), he may require the Minister to refer that decision to a New Drug Committee and thereupon shall provide the Minister with a statement of the reasons for his dissatisfaction and any information and material upon which he relies in support of those reasons.
(3) Where the Minister has been required to refer a decision to a New Drug Committee pursuant to subsection (2), he shall appoint a member of the New Drug Committee, the dissatisfied manufacturer shall appoint a member of the New Drug Committee and the two members so appointed shall appoint a third member of the New Drug Committee who shall be chairman, or, if they are unable to do so within a reasonable time, the Minister shall appoint a third member of the New Drug Committee who shall be chairman.
(4) Any person who is in the full-time employment of the Department or in the full-time employment of the dissatisfied manufacturer shall not be appointed a member of a New Drug Committee.
(4.1) A member of a New Drug Committee shall, on appointment, sign an undertaking not to disclose or use any information, material, data, evidence or representations considered pursuant to subsection (6).
(5) The Minister shall pay the reasonable fees and costs incurred by the member of the New Drug Committee appointed by the Minister, and the dissatisfied manufacturer shall pay the reasonable fees and costs incurred by the member appointed by the dissatisfied manufacturer, and the Minister and the dissatisfied manufacturer shall each pay half of the reasonable fees and costs incurred by the chairman.
(6) The New Drug Committee formed pursuant to subsection (3) shall consider the reasons for the decision of the Minister, the reasons for the dissatisfaction of the dissatisfied manufacturer and any information or material in support of the reasons of the Minister or the dissatisfied manufacturer and may consider other evidence, material, information or representations.
(7) The New Drug Committee formed pursuant to subsection (3) shall report its findings and recommendations to the Minister.
(7.1) No member of a New Drug Committee shall disclose or use any information, material, data, evidence or representations considered pursuant to subsection (6).
(8) Where the Minister has received the findings and recommendations of a New Drug Committee, he may reconsider the decision to which those findings and recommendations relate.
- SOR/95-411, s. 12
- SOR/2001-203, s. 9
- SOR/2011-88, s. 21
Pre-positioning of Designated COVID-19 Drugs
C.08.009.01 The following definitions apply in sections C.08.009.03 to C.08.009.05.
- Chief Public Health Officer
Chief Public Health Officer means the Chief Public Health Officer appointed under subsection 6(1) of the Public Health Agency of Canada Act. (administrateur en chef de la santé publique)
- foreign regulatory authority
foreign regulatory authority means a government agency or other entity outside Canada that has a legal right to control the manufacturing, use or sale of drugs within its jurisdiction and that may take enforcement action to ensure that drugs marketed within its jurisdiction comply with the applicable legal requirements. (autorité réglementaire étrangère)
C.08.009.02 Sections C.08.009.03 to C.08.009.05 apply in respect of a designated COVID-19 drug if the following conditions are met:
(a) a notice of compliance has not been issued under section C.08.004 or C.08.004.01 in respect of the designated COVID-19 drug; and
(b) Her Majesty in right of Canada has entered into a contract for the procurement of the designated COVID-19 drug.
C.08.009.03 (1) The holder of an establishment licence may import a designated COVID-19 drug if the following conditions are met:
(a) the Chief Public Health Officer provides the Minister with
(i) information indicating that
(A) the designated COVID-19 drug is the subject of a new drug submission that was filed under section C.08.002, or
(B) an application has been submitted to a foreign regulatory authority to authorize the sale of the designated COVID-19 drug,
(ii) the name of the designated COVID-19 drug and a description of it,
(iii) the name and contact information of the designated COVID-19 drug’s manufacturer,
(iv) information specifying the quantity of the designated COVID-19 drug to be imported,
(v) the name and contact information of any holder of an establishment licence who is proposed to be an importer of the designated COVID-19 drug, and
(vi) the civic address of the place where the designated COVID-19 drug will be stored after importation;
(b) the holder provides the Minister with
(i) the name and contact information of each fabricator, packager/labeller and tester of the designated COVID-19 drug and the civic address of each building at which the designated COVID-19 drug will be fabricated, packaged/labelled or tested, specifying for each building
(A) the activities referred to in Table I to section C.01A.008 that apply to the designated COVID-19 drug,
(B) the categories referred to in Table II to that section that apply to the designated COVID-19 drug, and
(C) for each category, the dosage form classes, if any, and whether the designated COVID-19 drug will be in a sterile form, and
(ii) a certificate from an inspector indicating that each fabricator’s, packager/labeller’s and tester’s buildings, equipment, practices and procedures meet the applicable requirements of Divisions 2 to 4 or, alternatively, other evidence establishing that those requirements are met; and
(c) the holder is one of those specified in the information that the Chief Public Health Officer provides under subparagraph (a)(v).
(2) Paragraph (1)(b) does not apply to the holder of an establishment licence in respect of a building referred to in subparagraph (1)(b)(i) if
(a) the building is listed in the licence; and
(b) the information referred to in clauses (1)(b)(i)(A) to (C) that the holder submitted in respect of the building in their application for the licence under section C.01A.005 or in an application to amend the licence under section C.01A.006, has not changed.
(3) If the conditions set out in subsection (1) are met, the Minister shall send a letter to the Chief Public Health Officer to that effect.
C.08.009.04 Sections A.01.040 and C.01.004.1, subsection C.01A.004(1), section C.01A.006 and Divisions 2 to 4, other than the following provisions, do not apply in respect of the importation, under section C.08.009.03, of a designated COVID-19 drug by the holder of an establishment licence:
(a) sections C.02.003.1, C.02.004 and C.02.006, as they apply to the storage of the designated COVID-19 drug by the holder;
(b) subsection C.02.012(1);
(c) sections C.02.013 and C.02.014;
(d) section C.02.015, as it applies to the storage and transportation of the designated COVID-19 drug by the holder;
(e) subsection C.02.021(1), as it applies to the storage of the designated COVID-19 drug by the holder;
(f) subsection C.02.022(1);
(g) section C.02.023;
(h) subsection C.02.024(1);
(i) section C.03.013; and
(j) section C.04.001.1, as it applies to the storage of the designated COVID-19 drug by the holder.
C.08.009.05 Despite anything in these Regulations, the holder of an establishment licence may distribute a designated COVID-19 drug that they have imported under section C.08.009.03 if the following conditions are met:
(a) the Chief Public Health Officer provides the Minister with the name of the designated COVID-19 drug and the civic address of the place where it will be stored after the distribution; and
(b) the designated COVID-19 drug is distributed to a person who will store it at that place.
Disclosure of Information in Respect of Clinical Trials
C.08.009.1 (1) In sections C.08.009.2 and C.08.009.3, information in respect of a clinical trial means information in respect of a clinical trial, within the meaning of section C.05.001, that is contained in a new drug submission, an extraordinary use new drug submission, an abbreviated new drug submission or an abbreviated extraordinary use new drug submission for a new drug for human use filed under section C.08.002, C.08.002.01 or C.08.002.1 or in a supplement to any of those submissions filed under section C.08.003.
(2) For greater certainty, the definition information in respect of a clinical trial includes information that is contained in a submission or supplement referred to in that definition and that is in respect of clinical testing involving human subjects in regards to which an application was filed under this Division before September 1, 2001.
C.08.009.2 (1) Information in respect of a clinical trial that is confidential business information ceases to be confidential business information when one of the following circumstances occurs with respect to the submission or supplement:
(a) the Minister issues a notice of compliance under section C.08.004 or C.08.004.01;
(b) in the case where the Minister issues a notice to the manufacturer under paragraph C.08.004(1)(b) or C.08.004.01(1)(b) and the manufacturer does not amend the submission or supplement under subsection C.08.004(2) or C.08.004.01(2), the applicable period referred to in the relevant subsection expires;
(c) the Minister issues a notice to the manufacturer under paragraph C.08.004(3)(b) or C.08.004.01(3)(b).
(2) Subsection (1) does not apply to information in respect of a clinical trial that
(a) was not used by the manufacturer in the submission or supplement to support the proposed conditions of use for the new drug or the purpose for which the new drug is recommended; or
(b) describes tests, methods or assays that are used exclusively by the manufacturer.
C.08.009.3 The Minister may disclose, without notifying the person to whose business or affairs the information relates or obtaining their consent, any information in respect of a clinical trial that has ceased to be confidential business information.
Sale of New Drug for Emergency Treatment
- SOR/2020-212, s. 1(F)
C.08.010 (1) The Minister may issue a letter of authorization to a manufacturer of a new drug authorizing the sale of a specified quantity of the new drug for human or veterinary use to a practitioner, for use in the emergency treatment of an animal or a person under the care of that practitioner, if
(a) the practitioner provides the following information to the Minister:
(i) the name of the new drug and details concerning the medical emergency for which the new drug is required,
(ii) the quantity of the new drug that is required,
(iii) subject to subsection (2), the information in the possession of the practitioner in respect of the use, safety and efficacy of the new drug,
(iv) the name and the civic address of the person to whom the new drug is to be shipped, and
(v) any other information the Minister may request to enable the Minister to determine whether to issue the letter of authorization;
(b) the practitioner agrees to
(i) provide a report to the manufacturer of the new drug and to the Minister describing the results obtained following the use of the new drug to address the medical emergency, including information respecting any adverse drug reactions observed by the practitioner, and
(ii) account to the Minister, on request, for all quantities of the new drug received;
(c) in the case of a new drug for human use, the person referred to in subparagraph (a)(iv) is a practitioner or a pharmacist; and
(d) in the case of a new drug for veterinary use, the person referred to in subparagraph (a)(iv) is a practitioner, a pharmacist or a person who may sell a medicated feed pursuant to section C.08.012.
(2) Subparagraph (1)(a)(iii) does not apply if the following conditions are met:
(a) the sale of the new drug has been authorized under subsection (1) to address the same medical emergency on at least one previous occasion;
(b) the European Medicines Agency or the United States Food and Drug Administration has authorized the sale of the new drug without terms or conditions, for the same use in its jurisdiction; and
(c) the Minister has not cancelled the assignment of a drug identification number for the new drug under paragraph C.01.014.6(2)(b) or (c) or subsection C.01.014.6(3).
(3) [Repealed, SOR/2021-271, s. 4]
(4) The letter of authorization must contain the following information:
(a) the name of the practitioner to whom the new drug may be sold;
(b) the name and the civic address of the person to whom the new drug may be shipped;
(c) the name of the new drug and the medical emergency in respect of which it may be sold; and
(d) the quantity of the new drug that may be sold to the practitioner to address the medical emergency.
(5) For the purposes of this section, the practitioner is not required to know the identity of the animal or the person under the care of that practitioner at the time the letter of authorization is issued.
- SOR/2013-172, s. 11
- SOR/2018-69, ss. 31(E), 32(F)
- SOR/2020-212, s. 2
- SOR/2021-271, s. 4
C.08.011 (1) Despite section C.08.002, the manufacturer may sell a new drug in accordance with a letter of authorization issued under subsection C.08.010(1).
(2) In the case of the sale under subsection (1) of a new drug for veterinary use that contains an active pharmaceutical ingredient set out in List A and that was not imported under section C.08.011.2, an annual report identifying the total quantity of the new drug that was sold, including an estimate of the quantity sold in respect of each animal species for which the drug is intended, must be submitted to the Minister by the manufacturer, if the new drug was present in Canada at the time of sale, otherwise by the practitioner.
(3) The sale of a new drug made in accordance with subsection (1) is exempt from the provisions of the Act and these Regulations other than this section.
(4) The annual report described in subsection (2) is for a period of one calendar year and must be submitted on or before March 31 of the year following the calendar year covered by the report, beginning with the first full calendar year during which the drug is first sold.
C.08.011.1 (1) The Minister may issue a letter of authorization to the manufacturer of a new drug authorizing the holder of an establishment licence to import a specified quantity of the new drug for human or veterinary use, if the following conditions are met:
(a) the manufacturer provides the following information to the Minister:
(i) the name of the new drug and details concerning the medical emergency for which the new drug will be imported,
(ii) the quantity of the new drug to be imported,
(iii) the name of the holder of an establishment licence who will import the new drug,
(iv) the civic address of the facility where the new drug is to be stored in Canada, and
(v) any other information the Minister may request to enable the Minister to determine whether to issue the letter of authorization;
(b) the establishment licence authorizes the importation of a new drug in the same category as the one to be imported; and
(c) the quantity that is to be imported does not exceed the amount that the Minister determines is likely to be required to address the medical emergency.
(2) [Repealed, SOR/2021-271, s. 5]
(3) The letter of authorization must contain the following information:
(a) the name of the new drug and the medical emergency in respect of which the letter is issued;
(b) the quantity of the new drug that may be imported to address the medical emergency;
(c) the name of the holder of an establishment licence who is authorized to import the new drug; and
(d) the civic address of the facility where the new drug is to be stored in Canada.
C.08.011.2 (1) Despite subsection C.01A.004(1), the holder of an establishment licence may import a new drug in accordance with a letter issued under subsection C.08.011.1(1).
(2) Section C.01A.006 and Divisions 2 to 4, except for the following provisions, do not apply to the importation of a new drug referred to in the letter of authorization:
(a) sections C.02.003.1 and C.02.004, as they apply to the storage of the new drug by the holder of an establishment licence;
(b) section C.02.006, as it applies to the storage of the new drug by the holder of an establishment licence;
(c) subsection C.02.012(1);
(d) sections C.02.013 and C.02.014;
(e) section C.02.015, as it applies to the storage and transportation of the new drug by the holder of an establishment licence;
(f) subsection C.02.021(1), as it applies to the storage of the new drug by the holder of an establishment licence;
(g) subsection C.02.022(1);
(h) section C.02.023;
(i) subsection C.02.024(1);
(j) section C.03.013; and
(k) section C.04.001.1, as it applies to the storage of the new drug by the holder of an establishment licence.
C.08.011.3 (1) Despite section C.08.002, the holder of an establishment licence who imports a new drug under section C.08.011.2 may distribute a new drug in accordance with a letter of authorization issued under subsection C.08.010(1).
(2) The holder of an establishment licence who distributes to a practitioner, in accordance with subsection (1), a new drug for veterinary use that contains an active pharmaceutical ingredient set out in List A must submit to the Minister an annual report identifying the total quantity of the new drug that was distributed, including an estimate of the quantity distributed in respect of each animal species for which the drug is intended.
(3) The distribution of a new drug made in accordance with subsection (1) is exempt from the provisions of the Act and these Regulations other than this section.
(4) The annual report described in subsection (2) is for a period of one calendar year and must be submitted on or before March 31 of the year following the calendar year covered by the report, beginning with the first full calendar year during which the drug is first distributed.
Sale of Medicated Feeds
C.08.012 (1) Notwithstanding anything in this Division, a person may sell, pursuant to a written prescription of a veterinary practitioner, a medicated feed if
(a) as regards the drug or drugs used as the medicating ingredient of the medicated feed,
(i) the Minister has assigned a drug identification number pursuant to section C.01.014.2, or
(ii) the sale is permitted by section C.08.005, C.08.011 or C.08.013;
(b) the medicated feed is for the treatment of animals under the direct care of the veterinary practitioner who signed the prescription;
(c) the medicated feed is for therapeutic purposes only; and
(d) the written prescription contains the following information:
(i) the name and address of the person named on the prescription as the person for whom the medicated feed is to be mixed,
(ii) the species, production type and age or weight of the animals to be treated with the medicated feed,
(iii) the type and amount of medicated feed to be mixed,
(iv) the proper name, or the common name if there is no proper name, of the drug or each of the drugs, as the case may be, to be used as medicating ingredients in the preparation of the medicated feed, and the dosage levels of those medicating ingredients,
(v) any special mixing instructions, and
(vi) labelling instructions including
(A) feeding instructions,
(B) a warning statement respecting the withdrawal period to be observed following the use of the medicated feed, and
(C) where applicable, cautions with respect to animal health or to the handling or storage of the medicated feed.
(2) For the purposes of this section, medicated feed has the same meaning as in the Feeds Regulations, 2024.
- SOR/80-741, s. 1
- SOR/92-130, s. 1
- SOR/93-202, s. 27
- SOR/2018-69, s. 27
- SOR/2024-132, s. 88
Experimental Studies
Conditions of Sale
C.08.013 (1) Notwithstanding anything in this Division, a person may sell a new drug proposed for use in animals to an experimental studies investigator in a quantity specified by the Minister for the purpose of conducting an experimental study in animals if
(a) the experimental studies investigator has been issued an experimental studies certificate pursuant to subsection C.08.015(1) and the certificate has not been suspended or cancelled pursuant to section C.08.018; and
(b) the drug is labelled in accordance with subsection C.08.016(1).
(2) For the purposes of this section and sections C.08.014 to C.08.018,
- experimental studies certificate
experimental studies certificate means a certificate issued pursuant to subsection C.08.015(1); (certificat d’études expérimentales)
- experimental studies investigator
experimental studies investigator means a person named as the investigator in an experimental studies certificate; (expert en études expérimentales)
- experimental study
experimental study means a limited test of a new drug in animals carried out by an experimental studies investigator. (étude expérimentale)
- SOR/81-333, s. 1
- SOR/2018-69, s. 27
Experimental Studies Certificate
C.08.014 (1) For the purpose of obtaining an experimental studies certificate, an applicant shall submit to the Minister, in writing, the following information and material:
(a) the brand name of the new drug or the identifying name or code proposed for the new drug;
(b) the objectives and an outline of the proposed experimental study of the new drug;
(c) the species, number and production type of animals in respect of which the new drug is to be administered;
(d) the name and address of the manufacturer of the new drug;
(e) the address of the premises in which the experimental study is to be conducted;
(f) a description of the facilities to be used to conduct the experimental study;
(g) the name, address and qualifications of the proposed experimental studies investigator;
(h) the chemical structure, if known, and the relevant compositional characteristics of the new drug;
(i) the proposed quantity of the new drug to be used for the experimental study;
(j) the results of any toxicological or pharmacological studies that may have been conducted with the new drug;
(k) the written agreement referred to in subsection (2); and
(l) such other information and material as the Minister may require.
(2) Where a food-producing animal is involved in an experimental study, the applicant referred to in subsection (1) shall, for the purposes of obtaining an experimental studies certificate, obtain from the owner of the animals, or from a person authorized by the owner, a written agreement not to sell the animal or any products from it without prior authorization from the experimental studies investigator.
(3) The Minister may request the manufacturer of a new drug to submit to him samples of the new drug or of any ingredient of the drug and, in satisfactory form and manner, any other information that the Minister requests and where such samples or information are not submitted, the Minister may refuse to issue an experimental studies certificate.
- SOR/81-333, s. 1
- SOR/93-202, s. 28
- SOR/2018-69, s. 27
C.08.015 (1) Where, on receipt of the information and material submitted pursuant to section C.08.014, the Minister has determined that
(a) the applicant is qualified as an experimental studies investigator for the purposes of the proposed experimental study,
(b) the facilities for the conduct of the experimental study are adequate for the purposes of the proposed experimental study, and
(c) the proposed experimental study can be conducted without undue foreseeable risk to humans or animals,
the Minister shall issue an experimental studies certificate for the purposes of the proposed experimental study and shall specify therein the quantity of the new drug that may be sold to the experimental studies investigator.
(2) If, on receipt of the information and material submitted under section C.08.014, the Minister determines that the requirements of paragraphs (1)(a), (b) and (c) have not been met, the Minister shall refuse to issue an experimental studies certificate.
- SOR/81-333, s. 1
- SOR/2018-69, ss. 25, 27
- SOR/2022-197, s. 9
- SOR/2023-247, s. 6
Labelling
C.08.016 (1) The label of a new drug that is sold pursuant to section C.08.013 shall show
(a) the brand name of the new drug or the identifying name or code proposed for the new drug;
(b) a warning statement to the effect that the drug is for use only in an experimental study in animals;
(c) the lot number of the drug;
(d) the name and address of the manufacturer of the drug; and
(e) the name of the person to whom the drug has been supplied.
(2) Sections C.01.004, C.01.005 and C.01.014 do not apply to a drug that is sold pursuant to section C.08.013 and labelled in accordance with subsection (1).
- SOR/81-333, s. 1
- SOR/88-378, s. 2
- SOR/93-202, s. 29
Conditions of Experimental Study
C.08.017 An experimental studies investigator shall
(a) use the new drug only in accordance with the outline of the experimental study;
(b) report immediately to the Minister all serious adverse drug reactions associated with the use of the new drug;
(c) report promptly to the Minister, on request, the results of the experimental study;
(d) return to the manufacturer, on request, all quantities of the new drug not used in the experimental study;
(e) maintain all records of the experimental study for a period of at least two years after the conclusion of the study and, on request, make such records available to the Minister;
(f) report promptly to the Minister any known disposition of animals involved in the study or of any products from the animals that is contrary to the terms of the agreement referred to in subsection C.08.014(2); and
(g) account to the Minister, on request, for all quantities of the new drug received by him.
- SOR/81-333, s. 1
- SOR/2001-203, s. 10
- SOR/2018-69, s. 27
Suspension or Cancellation of Experimental Studies Certificate
C.08.018 (1) If the Minister determines that it is necessary in order to safeguard animal health or public health or to promote public safety, he or she may suspend for a definite or indefinite period or cancel an experimental studies certificate.
(2) Without limiting the generality of subsection (1), the Minister may suspend or cancel an experimental studies certificate if
(a) the information and material submitted pursuant to section C.08.014 contains an untrue statement or contains any omission concerning the properties of the drug that were known or ought reasonably to have been known to the manufacturer or the experimental studies investigator;
(b) the labelling of the new drug is, at any time, false, misleading, deceptive or incomplete;
(c) the qualifications of the experimental studies investigator prove to be inadequate;
(d) there is evidence that the experimental studies investigator has not complied with the conditions referred to in section C.08.017; or
(e) an action of the manufacturer in respect of the new drug has resulted in his conviction for a violation of section C.08.002.
- SOR/81-333, s. 1
- SOR/2018-69, ss. 26, 27
DIVISION 9
Non-prescription Drugs
C.09.001 This Division does not apply to
(a) a drug that is required by these Regulations or the Narcotic Control Regulations to be sold only on prescription; or
(b) a drug for use exclusively in animals.
- SOR/84-145, s. 4
Analgesics
General
C.09.010 No manufacturer or importer shall, after June 30, 1986, sell a drug for analgesia that contains a combination of
(a) a salt or derivative of salicylic acid with another salt or derivative of salicylic acid or with salicylamide; or
(b) acetaminophen with a salt or derivative of salicylic acid or with salicylamide.
- SOR/84-145, s. 4
C.09.011 Each label of a drug that is intended for internal use and contains acetaminophen, salicylic acid or a salt or derivative thereof shall, after June 30, 1986, carry a caution
(a) to consult a physician if the underlying condition requires continued use for more than five days; and
(b) that it is hazardous to exceed the maximum recommended dose unless advised by a physician.
- SOR/84-145, s. 4
- SOR/86-589, s. 1
C.09.012 Each label of a drug that is intended for internal use and contains salicylic acid or a salt or derivative thereof shall after June 30, 1986, carry a warning statement to consult a physician before taking the drug during the last three months of pregnancy or when nursing.
- SOR/84-145, s. 4
Acetaminophen
C.09.020 (1) The adult standard dosage unit of acetaminophen shall be 325 mg.
(2) The children’s standard dosage units of acetaminophen shall be 80 mg or 160 mg.
- SOR/84-145, s. 4
- SOR/90-587, s. 4
C.09.021 (1) In this Division, acetaminophen product means a drug that contains
(a) acetaminophen as a single medicinal ingredient; or
(b) acetaminophen in combination with caffeine.
(2) No manufacturer or importer shall sell an acetaminophen product unless it meets the requirements of this Division.
(3) [Repealed, SOR/90-587, s. 5]
- SOR/84-145, s. 4
- SOR/90-587, s. 5
C.09.022 (1) Subject to subsections (2) to (4), an acetaminophen product sold in the form of a tablet, capsule or other solid dosage form intended for oral administration shall contain one adult standard dosage unit of acetaminophen per individual dosage form.
(2) An acetaminophen product in the form of a tablet, capsule or other solid dosage form intended for oral administration may contain 500 mg of acetaminophen per individual dosage form if it has a label that states that it is not a standard dosage unit product.
(3) An acetaminophen product sold in the form of a tablet, capsule or other solid dosage form that is intended for oral administration may contain 325 mg of acetaminophen for immediate release and another 325 mg for subsequent release, if it has a label that states that it is not a standard dosage unit product.
(4) An acetaminophen product sold in the form of a tablet, capsule or other solid dosage form that is intended for oral administration and that is specially recommended for children shall contain one children’s standard dosage unit of acetaminophen per individual dosage form.
(5) An acetaminophen product in the form of a liquid that is intended to be taken as drops and that is specially recommended for children shall contain one children’s standard dosage unit of acetaminophen per millilitre of the product.
(6) A package of an acetaminophen product described in subsection (5) shall be accompanied by a measuring device capable of accurately delivering 0.5 mL of the product.
(7) An acetaminophen product in the form of a liquid that is not intended to be taken as drops and that is specially recommended for children shall contain one children’s standard dosage unit per teaspoon of the product.
(8) An acetaminophen product in the form of a liquid shall contain one adult standard dosage unit of acetaminophen per teaspoon of the product.
- SOR/84-145, s. 4
- SOR/85-966, s. 4
- SOR/86-954, s. 1
- SOR/99-441, s. 1
Salicylates
C.09.030 (1) The adult standard dosage unit of a salicylate shall be
(a) in the case of acetylsalicylic acid, sodium salicylate and magnesium salicylate, 325 mg; and
(b) in the case of choline salicylate, 435 mg.
(2) The children’s standard dosage unit of a salicylate shall be
(a) in the case of acetylsalicylic acid, sodium salicylate and magnesium salicylate, 80 mg; and
(b) in the case of choline salicylate, 110 mg.
- SOR/84-145, s. 4
C.09.031 (1) In this Division, salicylate product means a drug that contains
(a) a salt or derivative of salicylic acid as a single medicinal ingredient;
(b) a salt or derivative of salicylic acid in combination with caffeine;
(c) a salt or derivative of salicylic acid in combination with one or more buffering agents or antacids; or
(d) a salt or derivative of salicylic acid in combination with caffeine and one or more buffering agents or antacids.
(2) No manufacturer or importer shall sell a salicylate product after June 30, 1986 unless it meets the requirements of this Division.
(3) No manufacturer or importer shall, until June 30, 1986, sell a salicylate product in a dosage unit other than one mentioned in this Division, unless the salicylate product was legally available for sale in Canada on February 1, 1984.
- SOR/84-145, s. 4
- SOR/85-966, s. 5(E)
C.09.032 (1) Subject to subsections (2) and (3) and section C.09.035, a salicylate product in the form of a tablet, capsule or other solid dosage form intended for oral administration shall contain one adult standard dosage unit of a salicylate per individual dosage form.
(2) A salicylate product in the form of a tablet, capsule or other solid dosage form intended for oral administration may contain
(a) 500 mg of acetylsalicylic acid, sodium salicylate or magnesium salicylate, or
(b) 670 mg of choline salicylate
per individual dosage form if it has a label that states that it is not a standard dosage unit product.
(3) A salicylate product in the form of a tablet, capsule or other solid dosage form intended for oral administration may contain
(a) two adult standard dosage units of a salicylate per individual dosage form if the label of the salicylate product states that each individual dosage form contains two adult standard dosage units; and
(b) three adult standard dosage units of a salicylate per individual dosage form if the label of the salicylate product states that each individual dosage form contains three adult standard dosage units.
- SOR/84-145, s. 4
- SOR/85-966, s. 6
C.09.033 (1) Subject to subsection (2), a salicylate product in the form of a liquid shall contain one adult standard dosage unit of a salicylate per teaspoon.
(2) A salicylate product in the form of a liquid may contain
(a) two adult standard dosage units of a salicylate per teaspoon if the label of the salicylate product states that each teaspoon of the product contains two adult standard dosage units; and
(b) three adult standard dosage units of a salicylate per teaspoon if the label of the salicylate product states that each teaspoon of the product contains three adult standard dosage units.
- SOR/84-145, s. 4
C.09.034 A salicylate product that is claimed to be buffered shall provide at least 1.9 milliequivalents of acid neutralizing capacity per adult standard dosage unit of a salicylate.
- SOR/84-145, s. 4
C.09.035 A salicylate product in the form of a tablet, capsule or other solid dosage form intended for oral administration and that is specially recommended for children shall contain one children’s standard dosage unit of a salicylate per individual dosage form.
- SOR/84-145, s. 4
DIVISION 10Access to Drugs in Exceptional Circumstances
C.10.001 (1) The following definitions apply in this section and in section C.10.002.
- foreign regulatory authority
foreign regulatory authority means a government agency or other entity outside Canada that has a legal right to control the manufacturing, use or sale of drugs within its jurisdiction. (autorité réglementaire étrangère)
- public health official
public health official means
(a) the Chief Public Health Officer appointed under subsection 6(1) of the Public Health Agency of Canada Act;
(b) the Chief Medical Officer of Health, or equivalent, of a province;
(c) the Surgeon General of the Canadian Armed Forces; or
(d) the Chief Medical Officer of Public Health for the Department of Indigenous Services. (responsable de la santé publique)
(2) Despite sections A.01.040 and C.01.004.1, any person who holds an establishment licence that authorizes the importation of a drug may import a drug for which a notice of compliance has not been issued under section C.08.004 or C.08.004.01, or for which a drug identification number has not been assigned under subsection C.01.014.2(1), if the following conditions are met:
(a) a public health official has, within the past year, notified the Minister in writing of
(i) an urgent public health need for the immediate use of the drug within their jurisdiction, and
(ii) the intended use or purpose of the drug;
(b) the drug is authorized by a foreign regulatory authority in the United States, Switzerland or the European Union to be sold for the same use or purpose as that described under subparagraph (a)(ii);
(c) the drug is in the same category as the category for which the licence was issued;
(d) the drug is imported directly from the country in which it is authorized to be sold by the foreign regulatory authority; and
(e) the drug is one for which the following information is set out in the List of Drugs for an Urgent Public Health Need that is published by the Government of Canada on its website, as amended from time to time:
(i) brand name,
(ii) medicinal ingredients,
(iii) dosage form,
(iv) strength,
(v) route of administration, and
(vi) identifying code or number, if any, assigned in the country in which the drug was authorized for sale.
(3) Sections C.01A.006 and C.01A.007 do not apply in respect of the importation of a drug under subsection (2).
(4) For greater certainty, a licensee may, despite subsection C.01A.004(1), import a drug under subsection (2) without having their licence amended under section C.01A.006.
(5) Divisions 2 to 4, other than the following provisions, do not apply to the importation of a drug under subsection (2):
(a) sections C.02.003.1 and C.02.004 as they apply to the storage of the drug by the licensee;
(b) section C.02.006;
(c) subsection C.02.012(1);
(d) sections C.02.013 and C.02.014;
(e) section C.02.015 as it applies to the storage and transportation of the drug by the licensee;
(f) subsection C.02.021(1) as it applies to the storage of the drug by the licensee;
(g) subsection C.02.022(1);
(h) section C.02.023;
(i) subsections C.02.024(1) and C.02.025(1);
(j) section C.03.013; and
(k) section C.04.001.1 as it applies to the storage of the drug by the licensee.
- SOR/2017-133, s. 2
- SOR/2023-18, s. 2
C.10.002 (1) A sale of a drug that is imported under subsection C.10.001(2) is exempt from the provisions of these Regulations only if the drug is sold to a person within the jurisdiction of a public health official who has notified the Minister as described in paragraph C.10.001(2)(a), for use in respect of the same urgent public health need for which it was imported.
(2) Any person who wholesales such a drug must hold an establishment licence to wholesale a drug in the same category and despite subsection (1), the following provisions apply in respect of the wholesale:
(a) sections C.02.003.1 and C.02.004 as they apply to the storage of the drug by the licensee;
(b) section C.02.006 as it applies to the storage of the drug by the licensee;
(c) subsection C.02.012(1);
(d) section C.02.013;
(d.1) section C.02.014;
(e) section C.02.015 as it applies to the storage and transportation of the drug by the licensee;
(e.1) subsection C.02.021(1) as it applies to storage;
(f) subsection C.02.022(1);
(f.1) [Repealed, SOR/2023-247, s. 7]
(g) section C.02.023; and
(h) subsection C.02.024(1).
- SOR/2017-133, s. 2
- SOR/2022-197, s. 10
- SOR/2023-247, s. 7
C.10.003 Every licensee who imports a drug under subsection C.10.001(2) must notify the Minister within 15 days after the day on which it is imported by providing the following information:
(a) the name, title and contact information of the person who imported the drug;
(b) the brand name of the drug;
(c) the medicinal ingredients, strength, dosage form and route of administration of the drug and any identifying code or number assigned to it in the country in which it was authorized for sale;
(d) the name of the country from which the drug was imported; and
(e) the total quantity of the drug imported.
- SOR/2017-133, s. 2
- SOR/2023-18, s. 3
C.10.004 (1) The following definitions apply in this section and in sections C.10.005 to C.10.011.
- designated drug
designated drug means a drug that is set out in the List of Drugs for Exceptional Importation and Sale. (drogue désignée)
- drug
drug means any of the following drugs for human use:
(a) drugs included in Schedule I, II, III, IV or V to the Controlled Drugs and Substances Act;
(b) prescription drugs;
(c) drugs that are listed in Schedule C or D to the Act; and
(d) drugs that are permitted to be sold without a prescription but that are to be administered only under the supervision of a practitioner. (drogue)
- foreign regulatory authority
foreign regulatory authority has the same meaning as in subsection C.10.001(1). (autorité réglementaire étrangère)
- List of Drugs for Exceptional Importation and Sale
List of Drugs for Exceptional Importation and Sale means the List of Drugs for Exceptional Importation and Sale that is published by the Government of Canada on its website, as amended from time to time. (Liste des drogues destinées aux importations et aux ventes exceptionnelles)
(2) In sections C.10.006 and C.10.009, batch certificate, fabricate, MRA country, package/label and recognized building have the same meanings as in subsection C.01A.001(1).
C.10.005 (1) The Minister may add a drug to the List of Drugs for Exceptional Importation and Sale only if the Minister has reasonable grounds to believe that
(a) there is a shortage or risk of shortage of another drug for which a notice of compliance has been issued under section C.08.004 or C.08.004.01 or for which a drug identification number has been assigned under subsection C.01.014.2(1); and
(b) the drug to be added to that list can be substituted for the drug referred to in paragraph (a).
(2) In subsection (1), shortage has the same meaning as in section C.01.014.8.
C.10.006 (1) A person who holds an establishment licence that authorizes the importation of a drug may import a designated drug if the following conditions are met:
(a) the licensee provides the Minister, electronically in a format specified by or acceptable to the Minister and not later than the third business day before the day on which the designated drug is imported, with a notification that contains the following information:
(i) the licensee’s name and contact information,
(ii) the name and contact information of each fabricator, packager/labeller and tester of the designated drug and the address of each building in which it is fabricated, packaged/labelled or tested,
(iii) in respect of the designated drug,
(A) its brand name,
(B) its medicinal ingredients,
(C) its dosage form,
(D) its strength,
(E) its route of administration,
(F) its identifying code or number, if any, assigned in the country in which it is authorized for sale, and
(G) a detailed description of its conditions of use,
(iv) the intended port of entry into Canada,
(v) the estimated date of arrival of the shipment of the designated drug, and
(vi) the total quantity of the designated drug that is intended to be imported on the date referred to in subparagraph (v);
(b) the designated drug is authorized to be sold by a foreign regulatory authority within its jurisdiction;
(c) the designated drug is in the same category as the category for which the establishment licence was issued;
(d) the following information is set out in the List of Drugs for Exceptional Importation and Sale in respect of the designated drug:
(i) the licensee’s name,
(ii) the information referred to in clauses (a)(iii)(A) to (F),
(iii) the name of the foreign regulatory authority referred to in paragraph (b), and
(iv) the date after which it may no longer be imported;
(e) the lot number of the designated drug is set out in the list referred to in paragraph (d), if applicable;
(f) the total quantity of the designated drug that the licensee imports does not exceed the maximum limit specified in the list referred to in paragraph (d) in respect of the drug, if applicable;
(g) the designated drug is imported on or before the date referred to in subparagraph (d)(iv); and
(h) the licensee has prepared a plan that specifies the measures to be taken in order for the licensee to comply with section C.10.011.
(2) In subsection (1), business day means a day other than
(a) a Saturday; or
(b) a Sunday or other holiday.
C.10.007 Sections A.01.040, A.01.044 and C.01.004.1 do not apply in respect of the importation, under section C.10.006, of a designated drug by a person who holds an establishment licence.
C.10.008 (1) Subject to sections C.10.009 and C.10.010, a sale of a designated drug that is imported under section C.10.006 is exempt from the following provisions:
(a) sections A.01.015, A.01.017 and A.01.051; and
(b) the provisions of Part C other than
(i) sections C.01.016, C.01.017, C.01.019 to C.01.020.1, C.01.040.3 to C.01.049.1 and C.01.051,
(ii) the provisions of Divisions 1A and 2, and
(iii) this section and sections C.10.009 to C.10.011.
(2) For greater certainty, for the purposes of section C.01.016, the manufacturer of a designated drug is required to comply only with the requirements set out in sections C.01.017 and C.01.019 in respect of the drug.
(3) Subsections (1) and (2) cease to apply in respect of the sale of a designated drug on its expiration date.
C.10.009 (1) Section C.02.019 does not apply to a person who holds an establishment licence in respect of a designated drug that they import under section C.10.006.
(2) The licensee shall perform the finished product testing on a sample of the designated drug that is taken either
(a) after receipt of each lot or batch of the designated drug on their premises in Canada; or
(b) before receipt of each lot or batch of the designated drug on their premises in Canada if the following conditions are met:
(i) the licensee has evidence satisfactory to the Minister to demonstrate that lots or batches of the designated drug sold to them by the vendor of the lot or batch are consistently manufactured in accordance with and consistently comply with the specifications for that drug, and
(ii) the designated drug has not been transported or stored under conditions that may affect its compliance with the specifications for that drug.
(3) In subsection (2), a reference to specifications is a reference to the specifications with which the designated drug is required to comply within the jurisdiction of the foreign regulatory authority referred to in paragraph C.10.006(1)(b).
(4) If the licensee receives on their premises in Canada a lot or batch of a designated drug whose useful life is more than 30 days, they shall visually inspect the lot or batch to confirm the identity of the product.
(5) Subsections (2) and (4) do not apply to the licensee if the designated drug is fabricated, packaged/labelled and tested in an MRA country at a recognized building and the following conditions are met:
(a) the address of the building is set out in their establishment licence; and
(b) they retain a copy of the batch certificate for each lot or batch of the designated drug that they receive for at least one year after the expiration date of the lot or batch.
(6) In this section, specifications has the same meaning as in section C.02.002.
C.10.010 (1) A person who holds an establishment license and who imports a designated drug under section C.10.006 is required to comply with paragraphs C.02.020(1)(a), (b) and (d) in respect of the drug but is not required to maintain the records referred to in those paragraphs on their premises in Canada.
(2) The Minister may request that the licensee provide to the Minister any of the records referred to in paragraphs C.02.020(1)(a), (b) or (d) in respect of the designated drug.
(3) The licensee shall provide the requested records electronically in a format specified by or acceptable to the Minister within the time limit specified by the Minister.
C.10.011 (1) A person who holds an establishment licence shall not sell a designated drug that they imported under section C.10.006 unless they ensure that the information referred to in clause C.10.006(1)(a)(iii)(G) is available in English and French and in a manner that permits the safe use of the drug.
(2) The licensee shall ensure that the information is available in accordance with subsection (1) until at least the end of the day on the latest expiration date of the designated drug that they imported.
DIVISION 11Public or Canadian Armed Forces Health Emergencies — Drugs for Immediate Use or Stockpiling
C.11.001 (1) The following definitions apply in this Division.
- foreign regulatory authority
foreign regulatory authority has the same meaning as in subsection C.10.001(1). (autorité réglementaire étrangère)
- initial public health official
initial public health official means the public health official named in an authorization issued under subsection C.11.003(1). (responsable de la santé publique initial)
- public health official
public health official means
(a) the Chief Public Health Officer appointed under subsection 6(1) of the Public Health Agency of Canada Act;
(b) the Chief Medical Officer of Health, or equivalent, of a province;
(c) the Medical Officer of Health, or equivalent, of a municipality;
(d) the Surgeon General of the Canadian Armed Forces; or
(e) the Chief Medical Officer of Public Health for the Department of Indigenous Services. (responsable de la santé publique)
- subsequent public health official
subsequent public health official means any public health official, other than the initial public health official, who obtains the quantity of a drug, or a portion of the quantity, that is specified in an authorization issued under subsection C.11.003(1). (responsable de la santé publique subséquent)
(2) This Division applies to a drug for human use in dosage form for which a drug identification number has not been assigned under subsection C.01.014.2(1) or for which a notice of compliance has not been issued under section C.08.004 or C.08.004.01, including drugs that have ceased to be considered to be natural health products by virtue of subsection 103.15(2) of the Natural Health Products Regulations.
C.11.002 (1) In order to address an actual, imminent or potential emergency, event or incident affecting public health or the health of members of the Canadian Armed Forces, a public health official may, on application to the Minister, obtain an authorization that permits a drug manufacturer to sell a specified quantity of a drug to the public health official, for immediate use or stockpiling or both.
(2) The application must
(a) set out the name of the public health official and include information setting out how they may be contacted at any time;
(b) set out the name of the manufacturer and include information setting out how they may be contacted at any time;
(c) describe the emergency, event or incident;
(d) state whether the drug is for immediate use or stockpiling or both;
(e) describe the use of the drug that is intended to address the emergency, event or incident;
(f) set out the civic address of the place to which the drug is to be shipped by the manufacturer;
(g) set out the following information about the drug:
(i) its brand name, if any, and either its proper name, common name and chemical name or its identifying name, code, number or mark,
(ii) its medicinal ingredients,
(iii) its strength,
(iv) its dosage form,
(v) the recommended dosage for the use described under paragraph (e),
(vi) its recommended route of administration,
(vii) the indications that have been approved by any foreign regulatory authority, if applicable,
(viii) its contraindications,
(ix) a summary of its safety profile, and
(x) the recommended storage conditions for the drug;
(h) specify the quantity of the drug required to address the emergency, event or incident;
(i) include a statement by the public health official, accompanied by supporting information or documents, attesting that
(i) there is an actual, imminent or potential emergency, event or incident affecting public health or the health of the members of the Canadian Armed Forces that is likely to result, in humans, in a serious or life-threatening disease, disorder or abnormal physical state,
(ii) immediate action is or would likely be required to diagnose, treat, mitigate or prevent the disease, disorder or abnormal physical state or its symptoms,
(iii) conventional therapies, if any, have failed, are unsuitable or are unavailable in Canada at the time the application is made, and
(iv) the known and potential benefits associated with the use of the drug described under paragraph (e) outweigh the known and potential risks associated with that use;
(j) include any information or document available to the public health official concerning the safety, efficacy and quality of the drug in respect of the use described under paragraph (e), including information published in a medical or scientific journal; and
(k) set out the following information, if known by the public health official:
(i) the names of the foreign regulatory authorities that have authorized the sale of the drug in their jurisdictions for the same use as that described under paragraph (e),
(ii) the names of the foreign regulatory authorities that have received an application for authorization to sell the drug in their jurisdictions for the use described under paragraph (e) but that have not yet made a decision in respect of that application at the time the public health official makes the application under subsection (1), and
(iii) the names of the foreign regulatory authorities that have refused to authorize the sale of the drug in their jurisdictions for any use, as well as the reason for the refusal.
(3) The public health official must provide the Minister with any additional information or document that the Minister determines is necessary for the purpose of reviewing the application, by the date specified by the Minister.
C.11.003 (1) The Minister may, after review of the application, issue an authorization to a manufacturer authorizing the sale of a specified quantity of the drug to the public health official for the use described in the application.
(2) In reviewing the application, the Minister must consider whether there is an alternative mechanism that would address the emergency, event or incident.
(3) The authorization must
(a) set out the date of issue;
(b) set out the name and contact information of the public health official;
(c) set out the name and contact information of the manufacturer;
(d) describe the emergency, event or incident;
(e) state whether the drug is for immediate use or stockpiling or both;
(f) describe the use for which the sale of the drug is authorized in order to address the emergency, event or incident;
(g) set out the civic address of the place to which the drug is to be shipped by the manufacturer;
(h) set out the following information about the drug:
(i) its brand name, if any, and either its proper name, common name and chemical name or its identifying name, code, number or mark,
(ii) its medicinal ingredients,
(iii) its strength,
(iv) its dosage form,
(v) its recommended dosage and route of administration, and
(vi) its recommended storage conditions; and
(i) specify the quantity of the drug that may be sold.
C.11.004 The initial public health official must notify the Minister, in writing, of any change to the information provided under paragraph C.11.002(2)(g), subparagraph C.11.002(2)(i)(iv), paragraph C.11.002(2)(j) or subsection C.11.002(3), within 30 days after the day on which they become aware of the change.
C.11.005 (1) Subject to subsection C.11.008(2), these Regulations, other than sections A.01.010, A.01.014 and A.01.045, subsections C.01.001(1) and (1.1) and this Division, do not apply to a drug that is sold in accordance with an authorization.
(2) In the case of a drug described in Schedule C or D of the Act, a drug that is sold in accordance with an authorization is exempt from the application of section 12 of the Act.
C.11.006 (1) The initial public health official must ensure that the drug bears a label or is accompanied by a document that clearly sets out the following information in English and French:
(a) the name and civic address of the drug’s manufacturer;
(b) a statement that the Minister has authorized the sale of the drug to address the emergency, event or incident described in the authorization;
(c) a statement that the drug is to be used only for the use described in the authorization;
(d) the drug’s brand name, if any, and either its proper name, common name and chemical name or its identifying name, code, number or mark;
(e) the drug’s medicinal ingredients;
(f) the drug’s strength;
(g) the drug’s dosage form;
(h) the drug’s recommended dosage and route of administration;
(i) the drug’s lot number, if known;
(j) all warnings and precautions in respect of the use of the drug, if any;
(k) the drug’s expiration date, or, if there is no expiration date, the stability testing date or the date on which the drug should be retested, as specified by the manufacturer;
(l) the drug’s recommended storage conditions; and
(m) the net contents of the drug’s container, in terms of the weight, volume, size or number of units of the drug in the container.
(2) Any subsequent public health official must ensure that the drug bears the label or is accompanied by the document.
(3) If the initial public health official becomes aware of a change to any of the information referred to in paragraph (1)(a), (h), (j), (k) or (l), they must
(a) ensure that the information on the drug’s label or in the accompanying document is updated; and
(b) notify of the change, in writing and without delay, any person to whom they have sold any quantity of the drug.
(4) If the person notified under paragraph (3)(b) is a subsequent public health official, they must notify of the change, in writing and without delay, any person to whom they have sold any quantity of the drug.
(5) Any person who is notified under paragraph (3)(b) or subsection (4) must ensure that the updated information accompanies, in writing, any quantity of the drug that remains in their possession.
C.11.007 (1) In addition to the information required under subsection C.11.006(1), the initial public health official or any subsequent public health official, as the case may be, must make the following information available to the following persons, in writing, in English and French:
(a) to the persons to whom the drug is administered and the persons who administer the drug, the known and potential benefits and risks associated with the use for which the sale of the drug is authorized, the recommended duration of use, if any, of the drug and instructions on how to report serious adverse drug reactions; and
(b) to the persons who administer the drug, the information referred to in paragraphs C.11.002(2)(a), (b) and (e) and subparagraphs C.11.002(2)(g)(v), (vii) and (viii), if that information is not set out on the drug’s label or accompanying document.
(2) If the initial public health official becomes aware of a change to any of the information referred to in subsection (1), they must, in writing and without delay, notify the relevant persons of the change.
C.11.008 (1) If the initial public health official is a person referred to in paragraph (d) of the definition public health official in subsection C.11.001(1), they must submit a written report to the Minister in respect of any serious adverse drug reaction to the drug, and include in the report the information referred to in paragraphs C.01.020.1(2)(b) to (l), no later than the 30th day after the day on which they become aware of the reaction.
(2) If the initial public health official is a person referred to in any of paragraphs (a) to (c) or (e) of the definition public health official in subsection C.11.001(1), the written report is not required and section C.01.020.1 applies in respect of the provision of information relating to serious adverse drug reactions.
C.11.009 (1) The initial public health official must monitor the response to the drug in the emergency, event or incident — including monitoring information they receive relating to serious adverse drug reactions — and must take reasonable steps to obtain information on that response.
(2) The initial public health official must, on request of the Minister, submit a written report to the Minister on the monitoring of the response to the drug in the emergency, event or incident during the time period specified by the Minister, as well as on any corrective measures taken as a result of the monitoring.
C.11.010 (1) The initial public health official must maintain all information about the sale and use of the drug in a way that allows them to submit the information, notices and reports referred to in sections C.11.004 and C.11.008 and subsection C.11.009(2).
(2) The initial public health official and any subsequent public health official must maintain all information about the sale and use of the drug in a way that allows them to communicate with persons to whom the drug has been administered if the health of those persons may be endangered by its use.
C.11.011 The initial public health official or any subsequent public health official, as the case may be, must retain the information, notices and reports referred to in sections C.11.004 and C.11.008, subsection C.11.009(2) and section C.11.010, as applicable, for 15 years after the end of the period to which the information, notices and reports relate.
C.11.012 The initial public health official must account to the Minister for any unused quantity of stockpiled drug remaining in their possession at the end of the preceding calendar year by the January 30 that follows the first full calendar year during which the drug is stockpiled and then by January 30 of each subsequent year.
C.11.013 (1) The Minister may cancel an authorization if the Minister has reasonable grounds to believe that the drug presents a serious or imminent risk of injury to human health.
(2) If the Minister cancels an authorization, the initial public health official must, without delay, notify of the cancellation any person to whom they have directly distributed any quantity of the drug.
(3) These Regulations apply to any unused quantity of the drug as of the day on which the cancellation takes effect.
C.11.014 (1) The Minister may issue an authorization that permits an initial public health official who is a person referred to in paragraph (a), (d) or (e) of the definition public health official in subsection C.11.001(1) to sell a specified quantity of a stockpiled drug to a practitioner for use in the emergency treatment of a person under the care of that practitioner, if
(a) the manufacturer of the drug has been issued a letter of authorization under subsection C.08.010(1) that authorizes the sale of a specified quantity of the drug to that practitioner for the emergency treatment of that person; and
(b) the use of the drug specified in the letter of authorization is the same as the use described in the authorization issued to the manufacturer under subsection C.11.003(1).
(2) This Division, other than this section, does not apply to a drug sold in accordance with an authorization issued under subsection (1).
PART DVitamins, Minerals and Amino Acids
D.01.001 (1) In this Part,
- advertise
advertise means to advertise to the general public; (faire de la publicité)
- brand name
brand name means, with reference to a drug, the name, whether or not including the name of any manufacturer, corporation, partnership or individual, in English or French,
(a) that is assigned to the drug by its manufacturer,
(b) under which the drug is sold or advertised, and
(c) that is used to distinguish the drug; (marque nominative)
- common name
common name means, with reference to a salt or derivative of a vitamin, the name in English or French by which the salt or derivative is
(a) commonly known, and
(b) designated in scientific or technical journals; (nom usuel)
- human milk fortifier
human milk fortifier has the same meaning as in section B.25.001; (fortifiant pour lait humain)
- human milk substitute
human milk substitute has the same meaning as in section B.25.001; (succédané de lait humain)
- prepackaged product
prepackaged product means any food that is contained in a package in the manner in which it is ordinarily sold to, or used or purchased by, a person; (produit préemballé)
- reasonable daily intake
reasonable daily intake, in respect of a food named in an item in Column I of Schedule K, means the amount of that food set out in Column II of that item; (ration quotidienne normale)
- recommended daily intake
recommended daily intake[Repealed, SOR/2016-305, s. 61]
- supplemental ingredient
supplemental ingredient has the same meaning as in section B.01.001; (ingrédient supplémentaire)
- supplemented food
supplemented food has the same meaning as in section B.01.001; (aliment supplémenté)
- supplemented food facts table
supplemented food facts table has the same meaning as in subsection B.01.001(1); (tableau des renseignements sur les aliments supplémentés)
- Table of Daily Values
Table of Daily Values has the same meaning as in subsection B.01.001(1); (Tableau des valeurs quotidiennes)
- Table of Reference Amounts
Table of Reference Amounts has the same meaning as in subsection B.01.001(1); (Tableau des quantités de référence)
- testimonial
testimonial, with respect to a food or drug that is represented as containing a vitamin, mineral nutrient or mineral, means any dramatized or undramatized pictorial, written or oral representation as to the result that is, has been or may be produced by the addition to a person’s diet of that vitamin, mineral nutrient or mineral, as the case may be; (témoignage)
- weighted recommended nutrient intake
weighted recommended nutrient intake means, in respect of a vitamin or mineral nutrient, the amount of the vitamin or mineral nutrient set out in Table II to Division 1 and Table II to Division 2. (apport nutritionnel recommandé pondéré)
(2) For the purposes of this Part, a serving of stated size of a food shall be
(a) based on the food as offered for sale;
(b) in either of the following cases, the net quantity of the food in the package:
(i) if the quantity of food in the package can reasonably be consumed by one person at a single eating occasion, or
(ii) if the package contains less than 200% of the reference amount for the food; and
(c) in all other cases, the amount indicated for the food according to the criteria set out in column 3A of the Table of Reference Amounts.
(3) A serving of stated size of a food shall be expressed as follows:
(a) in the case of a single-serving prepackaged product to which paragraph (2)(b) applies, per package and using the following units:
(i) in grams, if the net quantity of the food is shown on the label by weight or by count, and
(ii) in millilitres, if the net quantity of the food is shown on the label by volume; and
(b) in the case of a multiple-serving prepackaged product to which paragraph (2)(c) applies, according to the following units set out in column 3B of the Table of Reference Amounts and according to the manner set out in that column:
(i) the household measure that applies to the product, and
(ii) the metric measure that applies to the product.
- SOR/88-559, s. 31
- SOR/93-202, s. 31
- SOR/96-259, s. 3
- SOR/2003-11, s. 27
- SOR/2016-305, s. 61
- SOR/2021-57, s. 17
- SOR/2022-169, s. 22
D.01.001.1 (1) The daily value of a vitamin or mineral nutrient set out in column 1 of Part 2 of the Table of Daily Values is, in respect of a food, the quantity
(a) set out in column 2, if the food is intended solely for infants six months of age or older but less than one year of age;
(b) set out in column 3, if the food is intended for infants six months of age or older but less than one year of age or for children one year of age or older but less than four years of age, and
(c) set out in column 4, in any other case.
(2) Subsection (1) does not apply if the food is
(a) a human milk fortifier; or
(b) a human milk substitute intended solely for infants less than six months of age.
- SOR/2016-305, s. 62
- SOR/2021-57, s. 18
D.01.001.2 If both a nutrition symbol, as defined in subsection B.01.001(1), and a statement or claim referred to in any of sections D.01.004 to D.01.007 and D.02.002 to D.02.005 appear on the principal display panel of a prepackaged product,
(a) the height of the upper case letters in the statement or claim must not exceed two times the height of the upper case letters, excluding any accents, in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”; and
(b) the height of the tallest ascender of the lower case letters in the statement or claim must not exceed two times the height of the tallest ascender of the lower case letters in the nutrition symbol, other than in the words “Health Canada” and “Santé Canada”.
DIVISION 1Vitamins in Foods
D.01.002 (1) In this Division, vitamin means any of the following vitamins:
(a) vitamin A;
(b) vitamin D;
(c) vitamin E;
(d) vitamin K;
(e) vitamin C;
(f) thiamin, thiamine or vitamin B1;
(g) riboflavin or vitamin B2;
(h) niacin;
(i) vitamin B6;
(j) folacin or folate;
(k) vitamin B12;
(l) pantothenic acid or pantothenate;
(m) biotin; and
(n) choline. (vitamine)
(2) For the purposes of this Division, no expression, other than an expression set out in subsection (1), shall be used to declare the vitamin content of a food.
(3) This Division applies only in respect of foods represented as containing a vitamin for use in human nutrition.
- SOR/88-559, s. 32
- SOR/2003-11, s. 28
- SOR/2016-305, s. 63
D.01.003 (1) For the purposes of these Regulations, the vitamin content of a food — other than a formulated liquid diet, human milk fortifier, human milk substitute or food represented as containing a human milk substitute — shall be determined
(a) in the case of vitamin A, in terms of the content of retinol and its derivatives and beta-carotene, calculated on the basis of micrograms of retinol activity equivalents (RAE) and expressed in micrograms on the basis of the following relationships:
(i) 1 RAE = 1 microgram of retinol, and
(ii) 1 RAE = 12 micrograms of beta-carotene;
(b) in the case of vitamin D, in terms of the content of cholecalciferol and ergocalciferol, expressed in micrograms;
(c) in the case of vitamin E, in terms of the content of d-alpha-tocopherol and dl-alpha-tocopherol and their derivatives, expressed in milligrams on the basis of the following relationships:
(i) one milligram d-alpha-tocopherol = one milligram vitamin E, and
(ii) one milligram dl-alpha-tocopherol = 0.74 milligram vitamin E;
(d) in the case of vitamin K, in terms of the content of phylloquinone and menaquinones, expressed in micrograms;
(e) in the case of vitamin C, in terms of the content of L-ascorbic acid and L-dehydroascorbic acid and their derivatives, calculated in milligram equivalents of L-ascorbic acid and expressed in milligrams;
(f) in the case of thiamin, thiamine or vitamin B1, and its derivatives, in terms of the content of thiamin, expressed in milligrams;
(g) in the case of riboflavin or vitamin B2 and its derivatives, in terms of the content of riboflavin, expressed in milligrams;
(h) in the case of niacin, in terms of the content of niacin and its derivatives, calculated in milligrams of nicotinic acid, plus the content of tryptophan, calculated in milligrams and divided by 60, with the total niacin equivalents (NE) expressed in milligrams;
(i) in the case of vitamin B6, in terms of the content of pyridoxine, pyridoxal and pyridoxamine and their derivatives, calculated in milligram equivalents of pyridoxine and expressed in milligrams;
(j) in the case of folate, in terms of the content of folic acid (pteroylmonoglutamic acid) and related compounds exhibiting the biological activity of folic acid, calculated on the basis of micrograms of dietary folate equivalents (DFE) and expressed in micrograms on the basis of the following relationships:
(i) 1 DFE = 1 μg food folate, and
(ii) 1 DFE = 0.6 μg folic acid from food with added folic acid;
(k) in the case of vitamin B12, in terms of the content of cyanocobalamin and related compounds exhibiting the biological activity of cyanocobalamin, calculated in microgram equivalents of cyanocobalamin and expressed in micrograms;
(l) in the case of pantothenic acid or pantothenate, in terms of the content of d-pantothenic acid, expressed in milligrams;
(m) in the case of biotin, in terms of the content of biotin, expressed in micrograms; and
(n) in the case of choline, in terms of the content of choline, expressed in milligrams.
(2) For the purpose of paragraph (1)(h), the content of tryptophan may be calculated
(a) where the protein originates from a food that contains protein from more than one source or from a source other than milk, meat, poultry, fish or eggs, as constituting 1.1 per cent of the protein;
(b) where the protein originates from milk, meat, poultry or fish, as constituting 1.3 per cent of the protein; and
(c) where the protein originates from eggs, as constituting 1.5 per cent of the protein.
- SOR/88-559, s. 32
- SOR/90-830, s. 7
- SOR/2016-305, s. 64
- SOR/2021-57, s. 19
- SOR/2022-197, s. 11
D.01.004 (1) It is prohibited, on the label of or in any advertisement for a food — other than a formulated liquid diet, human milk fortifier, human milk substitute or food represented as containing a human milk substitute — to make a statement or claim concerning the vitamin content of the food unless
(a) the vitamin is set out in column 1 of Part 2 of the Table of Daily Values;
(b) the percentage of the daily value of the vitamin, per serving of stated size, is 5% or more; and
(c) the vitamin content is declared on the label or in the advertisement as a percentage of the daily value, per serving of stated size.
(1.1) The condition set out in paragraph (1)(c) need not be met if the statement or claim described in subsection (1) is made on a label of or in any advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
(2) If a statement or claim described in subsection (1) is made in an advertisement for a food that is not a prepackaged product or in an advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the percentage of the daily value, per serving of stated size, shall,
(a) in the case of an advertisement, other than a radio or television advertisement, be
(i) adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once, and
(ii) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once;
(b) in the case of a radio advertisement or the audio portion of a television advertisement, immediately precede or follow the statement or claim; or
(c) in the case of a television advertisement, be communicated
(i) in the audio mode, if the statement or claim is made only in the audio portion of the advertisement or in both the audio and visual portions, or
(ii) in the audio or visual mode, if the statement or claim is made only in the visual portion of the advertisement.
(3) The percentage of the daily value, per serving of stated size, that is communicated in the visual mode of a television advertisement in accordance with subparagraph (2)(c)(ii) shall
(a) appear concurrently with and for at least the same amount of time as the statement or claim;
(b) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(c) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
(4) Paragraph (1)(b) does not apply in respect of a declaration of the vitamin content in a nutrition facts table or supplemented food facts table.
(5) Paragraph (1)(c) does not apply to a declaration of the biotin content as required by subparagraph B.24.202(a)(vi).
- SOR/84-300, s. 57(E)
- SOR/88-559, s. 32
- SOR/90-830, s. 8
- SOR/96-259, s. 9
- SOR/2003-11, s. 29
- SOR/2016-305, ss. 65, 75(F)
- SOR/2021-57, s. 20
- SOR/2022-169, s. 23
D.01.005 [Repealed, SOR/2003-11, s. 29]
D.01.006 No person shall, on the label of or in any advertisement for a food, make any claim concerning the action or effects of a vitamin contained in the food, except to the effect that the vitamin
(a) is a factor in the maintenance of good health; and
(b) is generally recognized as an aid in maintaining the functions of the body necessary to the maintenance of good health and normal growth and development.
- SOR/88-559, s. 32
D.01.007 (1) If a component of an ingredient of a prepackaged product set out in the table to subsection B.01.009(1) is a vitamin, no person shall, on the label of or in any advertisement for the prepackaged product, make a statement or claim concerning the vitamin as a component of that ingredient unless
(a) despite subsection B.01.008.2(6), the vitamin is declared by its common name, and that common name is shown in parentheses immediately after the ingredient in respect of which it is a component, except that if a source of a food allergen or gluten is required by paragraph B.01.010.1(8)(a) to be shown immediately after that ingredient, the common name of the vitamin is instead shown immediately after that source; and
(b) all components of the ingredient are declared.
(2) Paragraph (1)(b) does not apply to flour used as an ingredient in the manufacture of a prepackaged product referred to in subsection (1).
- SOR/84-300, s. 59(E)
- SOR/88-559, s. 32
- SOR/2003-11, s. 30
- SOR/2011-28, s. 7
- SOR/2016-305, s. 66
D.01.008 (1) Sections D.01.009, D.01.010 and D.01.011 do not apply to a human milk fortifier.
(2) Sections D.01.009 and D.01.011 do not apply to a supplemented food.
D.01.009 Subject to section D.01.010, no person shall sell a food to which any of the following vitamins have been added unless a reasonable daily intake of that food by a person would result in the daily intake by such person of that vitamin in an amount not less than,
(a) in the case of vitamin A, 1,600 International Units;
(b) in the case of thiamine, 0.6 milligram;
(c) in the case of riboflavin, 1.0 milligram;
(d) in the case of niacin or niacinamide, six milligrams;
(e) in the case of ascorbic acid, 20 milligrams; and
(f) in the case of vitamin D, 300 International Units.
D.01.010 Where a food to which any of the following vitamins have been added is represented as being solely for use in the feeding of children under two years of age, no person shall sell such food unless a reasonable daily intake of that food by a child under two years of age would result in the daily intake by the child of that vitamin in an amount not less than,
(a) in the case of vitamin A, 1,000 International Units;
(b) in the case of thiamine, 0.4 milligram;
(c) in the case of riboflavin, 0.6 milligram;
(d) in the case of niacin or niacinamide, four milligrams;
(e) in the case of pyridoxine, 0.6 milligram;
(f) in the case of ascorbic acid, 20 milligrams;
(g) in the case of vitamin D, 300 International Units; and
(h) in the case of vitamin E, five International Units.
D.01.011 No person shall sell a food to which any of the following vitamins have been added if a reasonable daily intake of that food by a person would result in the daily intake by such person of that vitamin in an amount more than,
(a) in the case of vitamin A, 2,500 International Units;
(b) in the case of thiamine, two milligrams;
(c) in the case of riboflavin, three milligrams;
(d) in the case of niacin or niacinamide, 20 milligrams;
(e) in the case of pyridoxine, 1.5 milligrams;
(f) in the case of ascorbic acid, 60 milligrams;
(g) in the case of vitamin D, 400 International Units; and
(h) in the case of vitamin E, 15 International Units.
D.01.011.1 Sections D.01.009, D.01.010 and D.01.011 do not apply in respect of vitamin D in
(a) a food for which a standard is set out in Division 8 of Part B if the standard requires that the food contain vitamin D; or
(b) margarine or calorie-reduced margarine.
D.01.012 No person shall, in advertising a food that is represented as containing a vitamin or on a label of such food,
(a) give any assurance or guarantee of any kind with respect to the result that may be, has been or will be obtained by the addition of the vitamin to a person’s diet; or
(b) refer to, reproduce or quote any testimonial.
D.01.013 [Repealed, SOR/2003-11, s. 31]
TABLE I[Repealed, SOR/2016-305, s. 67]
TABLE II
Weighted Recommended Nutrient Intake
| Item | Column I | Column II | Column III |
|---|---|---|---|
| Vitamin | Units | Amount | |
| 1 | Biotin | micrograms | 90 |
| 2 | Folacin | micrograms | 195 |
| 3 | Niacin | niacin equivalents | 16 |
| 4 | Pantothenic Acid | milligrams | 5.0 |
| 5 | Riboflavin | milligrams | 1.2 |
| 6 | Thiamine | milligrams | 1.0 |
| 7 | Vitamin A | retinol equivalents | 870 |
| 8 | Vitamin B6 | milligrams | 1.0 |
| 9 | Vitamin B12 | micrograms | 1.0 |
| 10 | Vitamin C | milligrams | 34 |
| 11 | Vitamin D | micrograms | 3.0 |
| 12 | Vitamin E | milligrams | 7.0 |
- SOR/96-259, s. 5
DIVISION 2Mineral Nutrients in Foods
D.02.001 (1) In this Division, mineral nutrient means any of the following chemical elements, whether alone or in a compound with one or more other chemical elements:
(a) sodium;
(b) potassium;
(c) calcium;
(d) phosphorus;
(e) magnesium;
(f) iron;
(g) zinc;
(h) iodide;
(i) chloride;
(j) copper;
(k) fluoride;
(l) manganese;
(m) chromium;
(n) selenium;
(o) cobalt;
(p) molybdenum;
(q) tin;
(r) vanadium;
(s) silicon; and
(t) nickel. (minéral nutritif)
(2) This Division applies only in respect of foods that are represented as containing a mineral nutrient for use in human nutrition.
- SOR/88-559, s. 34
- SOR/90-830, s. 9(F)
D.02.002 (1) It is prohibited, on the label of or in any advertisement for a food — other than salt for table or general household use containing added iodide, prepackaged water and ice, a formulated liquid diet, human milk fortifier, human milk substitute or food represented as containing a human milk substitute — to make a statement or claim concerning the mineral nutrient content of the food unless
(a) the mineral nutrient is set out in column 1 of Part 2 of the Table of Daily Values;
(b) the percentage of the daily value of the mineral nutrient, per serving of stated size, is 5% or more; and
(c) the mineral nutrient content is declared on the label or in the advertisement as a percentage of the daily value, per serving of stated size.
(1.1) The condition set out in paragraph (1)(c) need not be met if the statement or claim described in subsection (1) is made on a label of or in any advertisement for a fresh vegetable or fruit or any combination of fresh vegetables or fruits without any added ingredients, an orange with added food colour or a fresh vegetable or fruit coated with mineral oil, paraffin wax, petrolatum or any other protective coating.
(2) If a statement or claim described in subsection (1) is made in an advertisement for a food that is not a prepackaged product or in an advertisement for a prepackaged product that is not made or placed by or on the direction of the manufacturer of the product, the percentage of the daily value, per serving of stated size, shall,
(a) in the case of an advertisement, other than a radio or television advertisement, be
(i) adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once, and
(ii) shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once;
(b) in the case of a radio advertisement or the audio portion of a television advertisement, immediately precede or follow the statement or claim; or
(c) in the case of a television advertisement, be communicated
(i) in the audio mode, if the statement or claim is made only in the audio portion of the advertisement or in both the audio and visual portions, or
(ii) in the audio or visual mode, if the statement or claim is made only in the visual portion of the advertisement.
(3) The percentage of the daily value, per serving of stated size, that is communicated in the visual mode of a television advertisement in accordance with subparagraph (2)(c)(ii) shall
(a) appear concurrently with and for at least the same amount of time as the statement or claim;
(b) be adjacent to, without any intervening printed, written or graphic material, the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once; and
(c) be shown in letters of at least the same size and prominence as those of the statement or claim, if the statement or claim is made only once, or the most prominent statement or claim, if the statement or claim is made more than once.
(4) Subsection (1) does not apply to a statement or claim made in respect of the sodium or potassium content.
(5) Paragraphs (1)(a) and (c) do not apply in respect of a declaration of the total fluoride ion content as required by sections B.12.002 and B.12.008.
(6) Paragraph (1)(b) does not apply in respect of a declaration of the mineral nutrient content in a nutrition facts table or supplemented food facts table.
(7) Paragraph (1)(c) does not apply in respect of a declaration of the chromium, copper, manganese, molybdenum and selenium content as required by subparagraph B.24.202(a)(v).
- SOR/84-300, s. 60(E)
- SOR/88-559, s. 34
- SOR/90-830, s. 10
- SOR/96-259, s. 9
- SOR/2003-11, s. 34
- SOR/2016-305, ss. 68, 75(F)
- SOR/2021-57, s. 22
- SOR/2022-169, s. 25
D.02.003 [Repealed, SOR/2003-11, s. 34]
D.02.004 No person shall, on the label of or in any advertisement for a food, make any claim concerning the action or effects of a mineral nutrient contained in the food, except to the effect that the mineral nutrient
(a) is a factor in the maintenance of good health; and
(b) is generally recognized as an aid in maintaining the functions of the body necessary to the maintenance of good health and normal growth and development.
- SOR/84-300, s. 61(E)
- SOR/88-559, s. 34
D.02.005 (1) If a component of an ingredient of a prepackaged product set out in the table to subsection B.01.009(1) is a mineral nutrient, no person shall, on the label of or in any advertisement for the prepackaged product, make a statement or claim concerning the mineral nutrient as a component of that ingredient unless
(a) despite subsection B.01.008.2(6), the mineral nutrient is declared by its common name, and that common name is shown in parentheses immediately after the ingredient in respect of which it is a component, except that if a source of a food allergen or gluten is required by paragraph B.01.010.1(8)(a) to be shown immediately after that ingredient, the common name of the mineral nutrient is instead shown immediately after that source; and
(b) all components of the ingredient are declared.
(2) Paragraph (1)(b) does not apply to flour used as an ingredient in the manufacture of a prepackaged product referred to in subsection (1).
- SOR/88-559, s. 34
- SOR/2003-11, s. 35
- SOR/2011-28, s. 8
- SOR/2016-305, s. 69
D.02.006 [Repealed, SOR/2003-11, s. 35]
D.02.007 [Repealed, SOR/88-559, s. 34]
D.02.008 No person shall, in advertising a food that is represented as containing a mineral nutrient or on a label of such food,
(a) give any assurance or guarantee of any kind with respect to the result that may be, has been or will be obtained by the addition of the mineral nutrient to a person’s diet; or
(b) refer to, reproduce or quote any testimonial.
D.02.009 (1) No person shall sell a food to which any of the following mineral nutrients have been added unless a reasonable daily intake of that food by a person would result in the daily intake by such person of that mineral nutrient in an amount not less than,
(a) in the case of calcium, 300 milligrams;
(b) in the case of phosphorus, 300 milligrams;
(c) in the case of iron, four milligrams; and
(d) in the case of iodine, 0.10 milligram.
(2) Subsection (1) does not apply to a human milk fortifier or a supplemented food.
D.02.010 (1) No person shall sell elemental iron powder for use in foods as a source of iron as a mineral nutrient unless
(a) subject to paragraph (b), the powder meets the specifications for
(i) Iron, Carbonyl,
(ii) Iron, Electrolytic, or
(iii) Iron, Reduced,
as set out in the Food Chemicals Codex, Third Edition, 1981, published by the National Academy of Sciences of the United States of America; and
(b) in the case of Iron, Reduced, 100 per cent by weight of the particles pass through a 100 mesh sieve and at least 95 per cent by weight of the particles pass through a 325 mesh sieve.
(2) No person shall sell a food to which elemental iron powder has been added as a source of iron as a mineral nutrient unless the powder meets the requirements referred to in paragraphs (1)(a) and (b).
- SOR/84-303, s. 1
D.02.011 No person shall sell a food to which sodium iron pyrophosphate has been added as a source of iron as a mineral nutrient unless
(a) the bioavailability of the iron in the food is not less than 50 per cent of the bioavailability of ferrous sulphate as determined by official method FO-42, Determination of Bioavailability of Iron, December 15, 1982; and
(b) that person retains documentary evidence showing that the bioavailability of the iron in the food has been determined by the official method referred to in paragraph (a) and, on request by the Minister, submits such evidence to the Minister.
- SOR/84-303, s. 1
- SOR/2018-69, s. 27
TABLE I[Repealed, SOR/2016-305, s. 70]
TABLE II
Weighted Recommended Nutrient Intake
| Item | Column I | Column II | Column III |
|---|---|---|---|
| Mineral Nutrient | Units | Amount | |
| 1 | Calcium | milligrams | 780 |
| 2 | Iodide | micrograms | 155 |
| 3 | Iron | milligrams | 10 |
| 4 | Phosphorus | milligrams | 885 |
| 5 | Magnesium | milligrams | 210 |
| 6 | Zinc | milligrams | 10 |
- SOR/96-259, s. 7
DIVISION 3Addition of Vitamins, Mineral Nutrients or Amino Acids to Foods
D.03.001 (1) In this Division, the expressions vitamin and mineral nutrient have the same meaning as in Divisions 1 and 2.
(2) This Division applies only in respect of foods that are represented as containing a vitamin, mineral nutrient or amino acid for use in human nutrition.
- SOR/88-559, s. 35
D.03.002 (1) Subject to section D.03.003, no person shall sell a food, other than a supplemented food, to which a vitamin, mineral nutrient or amino acid has been added unless the food is listed in Column I of the Table to this section and the vitamin, mineral nutrient or amino acid, as the case may be, is listed opposite that food in Column II.
(2) No milk or milk product or derivative listed in Column I of the Table to this section applies to the lacteal secretion obtained from the mammary gland of any animal other than a cow, genus Bos, or a product or derivative of such secretion unless that animal is identified therein.
TABLE
Column I Column II Food Vitamin, Mineral Nutrient or Amino Acid 1 Breakfast cereals Thiamine, niacin, vitamin B6, folic acid, pantothenic acid, magnesium, iron and zinc. 2 Fruit nectars, vegetable drinks, bases and mixes for vegetable drinks and a mixture of vegetable juices Vitamin C. 2.1 Fruit flavoured drinks that meet all the requirements of section B.11.150 Vitamin C, folic acid, thiamine, iron, potassium. 2.2 Bases, concentrates and mixes that are used for making fruit flavoured drinks and that meet all the requirements of section B.11.151 Vitamin C, folic acid, thiamine, iron, potassium. 3 Infant cereal products Thiamine, riboflavin, niacin or niacinamide, calcium, phosphorus, iron, iodine. 4 Margarine and other similar substitutes for butter Vitamin A, Vitamin D, alpha-tocopherol 5 Alimentary pastes Thiamine, riboflavin, niacin or niacinamide, folic acid, pantothenic acid, vitamin B6, iron, magnesium 6 Human milk fortifiers, infant formulas and formulated liquid diets - Amino acids — alanine, arginine, aspartic acid, cystine, glutamic acid, glycine, histidine, hydroxyproline, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, taurine, threonine, tryptophan, tyrosine, valine;
- Minerals — calcium, chloride, chromium, copper, iodide, iron, magnesium, manganese, molybdenum, phosphorus, potassium, selenium, sodium, zinc;
- Vitamins — alpha-tocopherol, biotin, choline, d-pantothenic acid, folic acid, niacin, riboflavin, thiamin, vitamin A, vitamin B6, vitamin B12, vitamin C, vitamin D, vitamin K.
6.1 Foods represented for use in a very low energy diet - Vitamins — alpha-tocopherol, biotin, d-pantothenic acid, folic acid, niacin, riboflavin, thiamine, vitamin A, vitamin B6, vitamin B12, vitamin C, vitamin D, vitamin K
- Minerals — calcium, chloride, chromium, copper, iodine, iron, magnesium, manganese, molybdenum, phosphorus, potassium, selenium, sodium, zinc
7 Flavoured beverage mixes and bases recommended for addition to milk Vitamin A, thiamine, niacin or niacinamide, vitamin C, iron. 8 Simulated meat products, simulated poultry meat products, meat product extenders and poultry product extenders Thiamine, riboflavin, niacin, pyridoxine, d-pantothenic acid, folic acid, vitamin B12, iron, magnesium, potassium, zinc, copper, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine. 9 Meal replacements and nutritional supplements - Vitamins — alpha-tocopherol, biotin, d-pantothenic acid, folic acid, niacin, riboflavin, thiamine, vitamin A, vitamin B6, vitamin B12, vitamin C, vitamin D
- Minerals — calcium, chloride, chromium, copper, iodine, iron, magnesium, manganese, molybdenum, phosphorus, potassium, selenium, sodium, zinc
9.1 Ready breakfast, instant breakfast and other similar breakfast replacement foods however described Vitamin A, thiamine, riboflavin, niacin or niacinamide, vitamin C, iron 10 Condensed milk, milk, milk powder, sterilized milk, (naming the flavour) milk Vitamin D. 11 Skim milk with added milk solids, partly skimmed milk with added milk solids, (naming the flavour) skim milk, (naming the flavour) partly skimmed milk, (naming the flavour) skim milk with added milk solids, (naming the flavour) partly skimmed milk with added milk solids, skim milk, partly skimmed milk, skim milk powder Vitamin A, vitamin D. 12 Evaporated milk Vitamin C, vitamin D. 13 Evaporated skim milk, concentrated skim milk, evaporated partly skimmed milk, concentrated partly skimmed milk Vitamin A, vitamin C, vitamin D. 14 Apple juice, reconstituted apple juice, grape juice, reconstituted grape juice, pineapple juice, reconstituted pineapple juice, apple and (naming the fruit) juice as described in section B.11.132, concentrated fruit juice except frozen concentrated orange juice Vitamin C. 15 Flour, White Flour, Enriched Flour or Enriched White Flour Thiamine, riboflavin, niacin, vitamin B6, folic acid, d-pantothenic acid, calcium, iron, magnesium. 16 [Repealed, SOR/94-689, s. 2] 17 Table salt, table salt substitutes Iodine. 18 Dehydrated potatoes Vitamin C. 19 Products simulating whole egg Vitamin A, thiamine, riboflavin, niacin or niacinamide, vitamin B6, d-pantothenic acid, folic acid, vitamin B12, alphatocopherol, calcium, iron, zinc, potassium. 20 [Repealed, SOR/90-830, s. 11] 21 Goat’s milk, goat’s milk powder Vitamin D 22 Partly skimmed goat’s milk, skimmed goat’s milk, partly skimmed goat’s milk powder, skimmed goat’s milk powder Vitamins A and D 23 Evaporated goat’s milk Vitamins C, D, folic acid 24 Evaporated partly skimmed goat’s milk, evaporated skimmed goat’s milk Vitamins A, C, D, folic acid 25 Pre-cooked rice as defined in subsection B.13.010.1(1) Thiamine, niacin, vitamin B6, folic acid, pantothenic acid, iron 26 Mineral water, spring water, water in sealed containers, prepackaged ice Fluorine 27 Liquid whole egg, dried whole egg, frozen whole egg, liquid yolk, dried yolk, frozen yolk, liquid egg-white, (liquid albumen), dried egg-white (dried albumen), frozen egg-white (frozen albumen), liquid whole egg mix, dried whole egg mix, frozen whole egg mix, liquid yolk mix, dried yolk mix, frozen yolk mix Vitamin A, Vitamin D, Vitamin E, thiamine, riboflavin, niacin, vitamin B6, folacin, vitamin B12, pantothenic acid, calcium, phosphorus, magnesium, potassium, iron, zinc
- SOR/78-64, s. 8
- SOR/78-403, s. 29
- SOR/78-478, s. 3
- SOR/78-637, s. 11(E)
- SOR/78-698, s. 10
- SOR/79-6, s. 1
- SOR/81-60, s. 14
- SOR/83-858, s. 2
- SOR/84-300, s. 62
- SOR/85-623, s. 4
- SOR/86-320, s. 2
- SOR/87-640, s. 10
- SOR/88-559, s. 36
- SOR/89-145, s. 3
- SOR/89-198, s. 18
- SOR/90-830, s. 11
- SOR/94-35, s. 5
- SOR/94-689, s. 2
- SOR/95-474, s. 6
- SOR/96-259, s. 8
- SOR/2010-143, s. 39(E)
- SOR/2021-57, s. 24
- SOR/2022-169, s. 27
D.03.003 Section D.03.002 does not apply to a food, other than a supplemented food, if all of the following conditions are met:
(a) the food is
(i) a gluten-free food referred to in paragraph B.24.003(1)(g), or
(ii) represented for a special dietary use referred to in paragraph B.24.003(1)(h) or (i);
(b) no standard is prescribed in these Regulations for the food; and
(c) the food is not advertised.
- SOR/78-64, s. 9
- SOR/84-334, s. 2
- SOR/90-830, s. 12
- SOR/95-444, s. 3
- SOR/2022-169, s. 28
DIVISION 4[Repealed, SOR/2003-196, s. 105]
DIVISION 5Minerals in Drugs
D.05.001 to D.05.007 [Repealed, SOR/2003-196, s. 106]
D.05.008 (1) Subject to subsection (2), no person shall sell a drug containing fluorine if the largest recommended daily dosage of that drug as shown on the label thereof would, if consumed by a person, result in a daily intake by that person of more than one milligram of fluoride ion.
(2) Subsection (1) does not apply to a drug sold by prescription.
- SOR/81-196, s. 2
D.05.009 Where a drug contains fluorine, both the inner and outer labels of the drug shall carry a cautionary statement that, if the drug is used in an area where the drinking water has a natural fluorine content in excess of 0.7 parts of fluoride ion per million parts of water or is artificially fluoridated, mottling of the tooth enamel of a user of the drug may result.
D.05.010 [Repealed, SOR/2003-196, s. 107]
PART ECyclamate Sweeteners
- SOR/2016-74, s. 11
E.01.001 (1) In this Part, cyclamate sweetener means any of the following substances sold as a sweetener:
(a) cyclohexyl sulfamic acid or any of its salts; and
(b) any substance containing cyclohexyl sulfamic acid or any of its salts.
(2) Part B does not apply to any cyclamate sweetener.
- SOR/78-422, s. 4
- SOR/2016-74, s. 12
Sale
E.01.002 No person shall sell a cyclamate sweetener that is not labelled as required by this Part.
- SOR/78-422, s. 4
- SOR/2016-74, s. 13
Advertising
E.01.003 No person shall, in advertising a cyclamate sweetener to the general public, make any representation other than with respect to the name, price and quantity of the sweetener.
- SOR/78-422, s. 4
- SOR/2016-74, s. 14
Labelling
E.01.004 Every cyclamate sweetener shall be labelled to state that it should be used only on the advice of a physician.
- SOR/78-422, s. 4
- SOR/2016-74, s. 15
E.01.005 Every cyclamate sweetener shall be labelled to show
(a) its energy value expressed, in calories, per teaspoon, drop, tablet or other measure used in the directions for use and per 100 grams or 100 millilitres; and
(b) a list of all its ingredients and, in the case of cyclohexyl sulfamic acid or any of its salts or a carbohydrate, its quantity.
- SOR/78-422, s. 4
- SOR/2016-74, s. 16
PART GControlled Drugs
DIVISION 1General
Definitions
Marginal note:Definitions
G.01.001 The following definitions apply in this Part.
- Act
Act means the Controlled Drugs and Substances Act. (Loi)
- advertisement
advertisement includes any representation by any means whatever for the purpose of promoting, directly or indirectly, the sale or other disposal of a controlled drug. (publicité)
- competent authority
competent authority means a public authority of a foreign country that is authorized under the laws of the country to approve the importation or exportation of controlled drugs into or from the country. (autorité compétente)
- compound
compound includes a preparation. (composé)
- controlled drug
controlled drug means
(a) a controlled substance set out in the schedule to this Part; or
(b) in respect of a midwife, nurse practitioner or podiatrist, a controlled substance set out in the schedule to this Part that the midwife, nurse practitioner or podiatrist may prescribe, possess or conduct an activity with, in accordance with sections 3 and 4 of the New Classes of Practitioners Regulations. (drogue contrôlée)
- designated criminal offence
designated criminal offence means
(a) an offence involving the financing of terrorism against any of sections 83.02 to 83.04 of the Criminal Code;
(b) an offence involving fraud against any of sections 380 to 382 of the Criminal Code;
(c) the offence of laundering proceeds of crime against section 462.31 of the Criminal Code;
(d) an offence involving a criminal organization against any of sections 467.11 to 467.13 of the Criminal Code; or
(e) a conspiracy or an attempt to commit, being an accessory after the fact in relation to, or any counselling in relation to, an offence referred to in any of paragraphs (a) to (d). (infraction désignée en matière criminelle)
- destroy
destroy, in respect of a controlled drug, means to alter or denature it to such an extent that its consumption is rendered impossible or improbable. (destruction)
- hospital
hospital means a facility that is
(a) licensed, approved or designated by a province in accordance with the laws of the province to provide care or treatment to persons or animals suffering from any form of disease or illness; or
(b) owned or operated by the Government of Canada or the government of a province and that provides health services. (hôpital)
- international obligation
international obligation means an obligation in respect of a controlled drug set out in a convention, treaty or other multilateral or bilateral instrument that Canada has ratified or to which Canada adheres. (obligation internationale)
- label
label has the same meaning as in section 2 of the Food and Drugs Act. (étiquette)
- licensed dealer
licensed dealer means the holder of a licence issued under section G.02.007. (distributeur autorisé)
- midwife
midwife has the same meaning as in section 1 of the New Classes of Practitioners Regulations. (sage-femme)
- nurse practitioner
nurse practitioner has the same meaning as in section 1 of the New Classes of Practitioners Regulations. (infirmier praticien)
- package
package includes anything in which a controlled drug is wholly or partly contained, placed or packed. (emballage)
- pharmacist
pharmacist means a person who is entitled under the laws of a province to practise pharmacy and who is practising pharmacy in that province. (pharmacien)
- podiatrist
podiatrist has the same meaning as in section 1 of the New Classes of Practitioners Regulations. (podiatre)
- preparation
preparation means a drug that contains a controlled drug and an active medicinal ingredient in a recognized therapeutic dose, other than a controlled drug. (préparation)
- prescription
prescription means an authorization given by a practitioner that a stated amount of a controlled drug be dispensed for the person named in it or the animal identified in it (ordonnance)
- qualified person in charge
qualified person in charge means the individual designated under subsection G.02.004(1). (responsable qualifié)
- Security Directive
Security Directive means the Directive on Physical Security Requirements for Controlled Substances and Drugs Containing Cannabis, as amended from time to time and published by the Government of Canada on its website. (Directive en matière de sécurité)
- senior person in charge
senior person in charge means the individual designated under section G.02.003. (responsable principal)
- test kit
test kit means a kit
(a) that contains a controlled drug and a reagent system or buffering agent;
(b) that is used during the course of a chemical or analytical procedure to test for the presence or quantity of a controlled drug for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose; and
(c) the contents of which are not intended or likely to be consumed by, or administered to, a person or an animal. (nécessaire d’essai)
- SOR/78-220, s. 1
- SOR/85-550, s. 1
- SOR/86-91, s. 1
- SOR/90-261, s. 1(F)
- SOR/92-386, s. 1
- SOR/97-228, s. 7
- SOR/97-515, s. 2
- SOR/2003-135, s. 2
- SOR/2004-238, s. 1
- SOR/2012-230, s. 6
- SOR/2018-147, s. 29
- SOR/2019-171, s. 1
Application
Marginal note:Agricultural implants
G.01.002 (1) The Act and this Part do not apply in respect of a controlled drug that is contained in an agricultural implant and set out in Part III of the schedule to this Part, but nothing in this section exempts such a drug from the requirements of Part C.
Marginal note:Definition of agricultural implant
(2) In this section, agricultural implant means a product that is presented in a form suitable to allow sustained release of an active ingredient over a certain period of time and that is intended for insertion under the skin of a food-producing animal for the purpose of increasing weight gain and improving feed efficiency.
- SOR/97-515, s. 3
- SOR/99-125, s. 1
- SOR/2003-34, s. 1
- SOR/2003-413, s. 1
- SOR/2018-69, ss. 67, 68
- SOR/2019-171, s. 1
G.01.002.1 [Repealed, SOR/2019-171, s. 1]
Marginal note:Member of police force
G.01.003 A member of a police force or a person acting under their direction and control who, in respect of the conduct of the member or person, is exempt from the application of subsection 4(2) or section 5, 6 or 7 of the Act by virtue of the Controlled Drugs and Substances Act (Police Enforcement) Regulations is, in respect of that conduct, exempt from the application of this Part.
- SOR/80-543, s. 11
- SOR/2004-238, s. 2(F)
- SOR/2019-171, s. 1
Marginal note:Application of Parts C and D
G.01.004 Except as otherwise provided in this Part, it is prohibited to sell or provide a controlled drug or a preparation that does not comply with all provisions of Parts C and D that are applicable to it.
- SOR/92-386, s. 2
- SOR/97-228, s. 8
- SOR/2019-171, s. 1
Possession
Marginal note:Authorized persons
G.01.005 (1) A person is authorized to possess a controlled drug set out in any of items 1 to 3, 8 to 10, 12 to 14, 16 and 17 of Part I of the schedule to this Part if the person has obtained the controlled drug in accordance with these Regulations, in the course of activities conducted in connection with the administration or enforcement of an Act or regulation, or from a person who is exempt under section 56 of the Act from the application of subsection 5(1) of the Act with respect to that controlled drug, and the person
(a) requires the controlled drug for their business or profession and is
(i) a licensed dealer,
(ii) a pharmacist, or
(iii) a practitioner who is registered and entitled to practise in the province in which they possess that drug;
(b) is a practitioner who is registered and entitled to practise in a province other than the province in which they have that possession for emergency medical purposes only;
(c) is a hospital employee or a practitioner in a hospital;
(d) has obtained the controlled drug for their own use
(i) from a practitioner, or
(ii) in accordance with a prescription that was not issued or obtained in contravention of these Regulations;
(e) is a practitioner of medicine who received the controlled drug under subsection G.06.003(1) or (2) and their possession is for the purpose of providing or delivering it to a person referred to in subsection G.06.003(3);
(f) is an agent or mandatary of a practitioner of medicine who received the controlled drug under subsection G.06.003(1) and their possession is for the purpose of providing or delivering it to a person referred to in subsection G.06.003(2);
(g) is employed as an inspector, a member of the Royal Canadian Mounted Police, a police constable, a peace officer or a member of the technical or scientific staff of the Government of Canada, the government of a province or a university in Canada and their possession is in connection with that employment;
(h) is not a practitioner of medicine referred to in paragraph (e) or an agent or mandatary referred to in paragraph (f), is exempted under section 56 of the Act with respect to possession of that controlled drug and their possession is for a purpose set out in the exemption; or
(i) is the Minister.
Marginal note:Agent or mandatary
(2) A person is authorized to possess a controlled drug referred to in subsection (1) if the person is acting as the agent or mandatary of any person who is authorized to possess it in accordance with any of paragraphs (1)(a) to (e), (h) and (i).
Marginal note:Agent or mandatary — person referred to in paragraph (1)(g)
(3) A person is authorized to possess a controlled drug referred to in subsection (1) if they
(a) are acting as the agent or mandatary of a person who they have reasonable grounds to believe is a person referred to in paragraph (1)(g); and
(b) possess the controlled drug for the purpose of assisting that person in the administration or enforcement of an Act or a regulation.
Test Kits
Marginal note:Authorized activities
G.01.006 A person may sell, possess or otherwise deal in a test kit if the following conditions are met:
(a) a registration number has been issued for the test kit under section G.01.008 and has not been cancelled under section G.01.009;
(b) the test kit bears, on its external surface,
(i) the name of the manufacturer,
(ii) the trade name or trademark, and
(iii) the registration number; and
(c) the test kit will be used for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose.
Marginal note:Application for registration number
G.01.007 (1) The manufacturer of a test kit may obtain a registration number for it by submitting to the Minister an application containing
(a) a detailed description of the design and construction of the test kit;
(b) a detailed description of the controlled drug and other substances, if any, contained in the test kit, including the qualitative and quantitative composition of each component; and
(c) a description of the proposed use of the test kit.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the person authorized by the applicant for that purpose; and
(b) include an attestation by that person that all of the information submitted in support of the application is correct and complete to the best of their knowledge.
Marginal note:Additional information or document
(3) The applicant must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance of registration number
G.01.008 On completion of the review of the application for a registration number, the Minister must issue a registration number for the test kit, preceded by the letters “TK”, if the Minister determines that the test kit will only be used for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose and that it contains
(a) a controlled drug and an adulterating or denaturing agent that are combined in such a manner and in such a quantity, proportion or concentration that the preparation or mixture has no significant drug abuse potential; or
(b) such small quantities or concentrations of any controlled drug as to have no significant drug abuse potential.
Marginal note:Cancellation of registration number
G.01.009 The Minister must cancel the registration number for a test kit if
(a) the test kit is removed from the market by the manufacturer;
(b) the Minister has reasonable grounds to believe that the test kit is used or is likely to be used for any purpose other than a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose; or
(c) the Minister has reasonable grounds to believe that the cancellation is necessary to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use.
G.01.010 [Repealed, SOR/2019-171, s. 1]
DIVISION 2Licensed Dealers
Authorized Activities
Marginal note:General
G.02.001 (1) A licensed dealer may produce, assemble, sell, provide, transport, send, deliver, import or export a controlled drug if they comply with this Part and the terms and conditions of their dealer’s licence and any permit issued under this Part.
Marginal note:Qualified person in charge present
(2) A licensed dealer may conduct an activity in relation to a controlled drug at their site only if the qualified person in charge or an alternate qualified person in charge is present at the site.
Marginal note:Permit — import and export
(3) A licensed dealer must obtain a permit to import or export a controlled drug.
Marginal note:Possession for export
(4) A licensed dealer may possess a controlled drug for the purpose of exporting it if they have obtained it in accordance with this Part.
- SOR/2004-238, s. 3
- SOR/2019-171, s. 1
G.02.001.1 [Repealed, SOR/2019-171, s. 1]
G.02.001.2 [Repealed, SOR/2019-171, s. 1]
Dealer’s Licences
Preliminary Requirements
Marginal note:Eligible persons
G.02.002 The following persons may apply for a dealer’s licence:
(a) an individual who ordinarily resides in Canada;
(b) a corporation that has its head office in Canada or operates a branch office in Canada; or
(c) the holder of a position that includes responsibility for controlled drugs on behalf of the Government of Canada, the government of a province, a police force, a hospital or a university in Canada.
G.02.002.1 [Repealed, SOR/2019-171, s. 1]
Marginal note:Senior person in charge
G.02.003 An applicant for a dealer’s licence must designate only one individual as the senior person in charge, who has overall responsibility for management of the activities with respect to controlled drugs that are specified in the licence application. The applicant may designate themself if the applicant is an individual.
- SOR/2004-238, s. 4
- SOR/2010-222, s. 2
- SOR/2012-230, s. 8
- SOR/2014-260, ss. 1, 16(F)
- SOR/2019-171, s. 1
G.02.003.1 [Repealed, SOR/2019-171, s. 1]
G.02.003.2 [Repealed, SOR/2019-171, s. 1]
G.02.003.3 [Repealed, SOR/2019-171, s. 1]
G.02.003.4 [Repealed, SOR/2019-171, s. 1]
G.02.003.5 [Repealed, SOR/2019-171, s. 1]
G.02.003.6 [Repealed, SOR/2019-171, s. 1]
G.02.003.7 [Repealed, SOR/2019-171, s. 1]
G.02.003.8 [Repealed, SOR/2019-171, s. 1]
G.02.003.9 [Repealed, SOR/2019-171, s. 1]
G.02.003.91 [Repealed, SOR/2019-171, s. 1]
Marginal note:Qualified person in charge
G.02.004 (1) An applicant for a dealer’s licence must designate only one individual as the qualified person in charge, who is responsible for supervising the activities with respect to controlled drugs that are specified in the licence application and for ensuring that those activities comply with this Part. The applicant may designate themself if the applicant is an individual.
Marginal note:Alternate qualified person in charge
(2) An applicant for a dealer’s licence may designate an individual as an alternate qualified person in charge, who is authorized to replace the qualified person in charge when that person is absent. The applicant may designate themself if the applicant is an individual.
Marginal note:Qualifications
(3) Only an individual who meets the following requirements may be designated as a qualified person in charge or an alternate qualified person in charge:
(a) they work at the site specified in the dealer’s licence;
(b) they
(i) are a person entitled or, if applicable, registered and entitled by a provincial professional licensing authority or a professional association in Canada and entitled to practise a profession that is relevant to their duties, such as pharmacist, practitioner, pharmacy technician or laboratory technician,
(ii) hold a diploma, certificate or credential awarded by a post-secondary educational institution in Canada in a field or occupation that is relevant to their duties, such as pharmacy, medicine, dentistry, veterinary medicine, pharmacology, chemistry, biology, pharmacy technician, laboratory technician, pharmaceutical regulatory affairs or supply chain management or security, or
(iii) hold a diploma, certificate or credential that is awarded by a foreign educational institution in a field or occupation referred to in subparagraph (ii) and hold
(A) an equivalency assessment as defined in subsection 73(1) of the Immigration and Refugee Protection Regulations, or
(B) an equivalency assessment issued by an organization or institution that is responsible for issuing equivalency assessments and is recognized by a province;
(c) they have sufficient knowledge of and experience with the use and handling of the controlled drugs specified in the dealer’s licence to properly carry out their duties; and
(d) they have sufficient knowledge of the provisions of the Act and this Part that are applicable to the activities specified in the dealer’s licence to properly carry out their duties.
Marginal note:Exception
(4) An applicant for a dealer’s licence may designate an individual who does not meet any of the requirements of paragraph (3)(b) as a qualified person in charge or an alternate qualified person in charge if
(a) no other individual working at the site meets those requirements;
(b) those requirements are not necessary for the activities specified in the licence; and
(c) the individual has sufficient knowledge — acquired from a combination of education, training or work experience — to properly carry out their duties.
- SOR/2004-238, s. 4
- SOR/2019-171, s. 1
Marginal note:Ineligibility
G.02.005 An individual is not eligible to be a senior person in charge, a qualified person in charge or an alternate qualified person in charge if, during the 10 years before the day on which the dealer’s licence application is submitted,
(a) in respect of a designated substance offence or a designated criminal offence or a designated offence as defined in subsection 2(1) of the Cannabis Act, the individual
(i) was convicted as an adult, or
(ii) was a young person who received an adult sentence, as those terms are defined in subsection 2(1) of the Youth Criminal Justice Act; or
(b) in respect of an offence committed outside Canada that, if committed in Canada, would have constituted a designated substance offence or a designated criminal offence or a designated offence as defined in subsection 2(1) of the Cannabis Act,
(i) the individual was convicted as an adult, or
(ii) if they committed the offence when they were at least 14 years old but less than 18 years old, the person received a sentence that was longer than the maximum youth sentence, as that term is defined in subsection 2(1) of the Youth Criminal Justice Act, that could have been imposed under that Act for such an offence.
Issuance of Licence
Marginal note:Application
G.02.006 (1) A person who intends to conduct an activity referred to in section G.02.001 must obtain a dealer’s licence for each site at which they intend to conduct activities by submitting an application to the Minister that contains the following information:
(a) if the licence is requested by
(i) an individual, the individual’s name,
(ii) a corporation, its corporate name and any other name registered with a province, under which it intends to conduct the activities specified in its dealer’s licence or by which it intends to identify itself, and
(iii) the holder of a position described in paragraph G.02.002(c), the applicant’s name and the title of the position;
(b) the municipal address, telephone number and, if applicable, the email address of the proposed site and, if different from the municipal address, its mailing address;
(c) the name, date of birth, telephone number and email address of the proposed senior person in charge;
(d) with respect to each of the proposed qualified person in charge and any proposed alternate qualified person in charge,
(i) their name, date of birth, telephone number and email address,
(ii) the title of their position at the site,
(iii) the name and title of the position of their immediate supervisor at the site,
(iv) if applicable, their profession they practise that is relevant to their duties, the name of the province that authorizes them to practise it and their authorization number, and
(v) their education, training and work experience that are relevant to their duties;
(e) the activities that are to be conducted and the controlled drugs in respect of which each of the activities is to be conducted;
(f) if the licence is requested to manufacture or assemble a product or compound that contains a controlled drug, other than a test kit, a list that includes, for each product or compound,
(i) the brand name of the product or the name of the compound,
(ii) the drug identification number that is assigned to the product under section C.01.014.2, if any,
(iii) the name of the controlled drug in the product or compound,
(iv) the strength per unit of the controlled drug in it, the number of units per package and the number of packages,
(v) if it is to be manufactured or assembled by or for another licensed dealer under a custom order, the name, municipal address and dealer’s licence number of the other licensed dealer, and
(vi) if the applicant’s name appears on the label of the product or compound, a copy of the inner label;
(g) if the licence is requested in order to produce a controlled drug other than a product or compound that contains a controlled drug,
(i) the name of the controlled drug,
(ii) the quantity that the applicant expects to produce under their licence and the period during which that quantity would be produced, and
(iii) if it is to be produced for another licensed dealer under a custom order, the name, municipal address and licence number of the other licensed dealer;
(h) if the licence is requested for an activity that is not described in paragraph (f) or (g), the name of the controlled drug for which the activity is to be conducted and the purpose of the activity;
(i) a detailed description of the security measures in place at the site, determined in accordance with the Security Directive; and
(j) a detailed description of the method for recording information that the applicant proposes to use for the purpose of section G.02.071.
Marginal note:Documents
(2) An application for a dealer’s licence must be accompanied by the following documents:
(a) if the applicant is a corporation, a copy of
(i) the certificate of incorporation or other constituting instrument, and
(ii) any document filed with the province in which its site is located that states its corporate name and any other name registered with the province under which the applicant intends to conduct the activities specified in its dealer’s licence or by which it intends to identify itself;
(b) individual declarations signed and dated by each of the proposed senior person in charge, and qualified person in charge and any proposed alternate qualified person in charge, attesting that the person is not ineligible for a reason specified in section G.02.005;
(c) a document issued by a Canadian police force in relation to each person referred to in paragraph (b), indicating whether, during the 10 years before the day on which the application is submitted, the person was convicted as specified in subparagraph G.02.005(a)(i) or received a sentence as specified in subparagraph G.02.005(a)(ii);
(d) if any of the persons referred to in paragraph (b) has ordinarily resided in a country other than Canada during the 10 years before the day on which the application is submitted, a document issued by a police force of that country indicating whether in that period that person was convicted as specified in subparagraph G.02.005(b)(i) or received a sentence as specified in subparagraph G.02.005(b)(ii);
(e) declaration, signed and dated by the proposed senior person in charge, attesting that the proposed qualified person in charge and any proposed alternate qualified person in charge have the knowledge and experience required under paragraphs G.02.004(3)(c) and (d); and
(f) if the proposed qualified person in charge or any proposed alternate qualified person in charge does not meet the requirement of subparagraph G.02.004(3)(b)(i), either
(i) a copy of the person’s diploma, certificate or credential referred to in subparagraph G.02.004(3)(b)(ii) or (iii), or
(ii) a detailed description of the education, training and work experience that is required under paragraph G.02.004(4)(c), together with supporting evidence, such as a copy of a course transcript or an attestation by the person who provided the training.
Marginal note:Signature and attestation
(3) The application must
(a) be signed and dated by the proposed senior person in charge; and
(b) include an attestation by that person that
(i) all information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the applicant.
Marginal note:Additional information and documents
(4) The applicant must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
G.02.007 Subject to section G.02.009, on completion of the review of the licence application, the Minister must issue a dealer’s licence, with or without terms and conditions, that contains
(a) the licence number;
(b) the name of the licensed dealer, their corporate name or the title of the position they hold;
(c) the activities that are authorized and the names of the controlled drugs in respect of which each activity may be conducted;
(d) the municipal address of the site at which the dealer may conduct the authorized activities;
(e) the security level at the site, determined in accordance with the Security Directive;
(f) the effective date of the licence;
(g) the expiry date of the licence, which must be not later than three years after its effective date;
(h) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected,
(ii) ensure conformity with the requirements associated with the security level that is referred to in paragraph (e), or
(iii) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use; and
(i) if the licensed dealer produces a controlled drug, the quantity that they may produce and the authorized production period.
Marginal note:Validity
G.02.008 A dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section G.02.027 or G.02.028.
Marginal note:Refusal
G.02.009 (1) The Minister must refuse to issue a dealer’s licence if
(a) the applicant may not apply for a licence under section G.02.002;
(b) during the 10 years before the day on which the licence application is submitted, the applicant has contravened
(i) a provision of the Act, the Cannabis Act or their regulations, or
(ii) a term or condition of a licence or permit issued to the applicant under any regulations made under the Act or issued to the applicant under the Cannabis Act or its regulations;
(c) during the 10 years before the day on which the application is submitted, the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subparagraph G.02.005(a)(i) or (b)(i) or received a sentence as specified in subparagraph G.02.005(a)(ii) or (b)(ii);
(d) an activity for which the licence is requested would contravene an international obligation;
(e) the applicant does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the licence is requested;
(f) the method referred to in paragraph G.02.006(1)(j) does not permit the recording of information as required under section G.02.071;
(g) the applicant has not complied with the requirements of subsection G.02.006(4) or the information and documents that they have provided are not sufficient to complete the review of the licence application;
(h) the Minister has reasonable grounds to believe that the applicant has submitted false or misleading information or false or falsified documents in or in support of the licence application;
(i) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the applicant has been involved in the diversion of a controlled drug to an illicit market or use or has been involved in an activity that contravened an international obligation; or
(j) the Minister has reasonable grounds to believe that the issuance of the licence would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to issue a licence under paragraph (1)(b) or (h) if the applicant meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the applicant does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the applicant has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to issue a licence, the Minister must send the applicant a notice that sets out the Minister’s reasons and gives the applicant an opportunity to be heard.
Renewal of Licence
Marginal note:Application
G.02.010 (1) To apply to renew a dealer’s licence, a licensed dealer must submit to the Minister an application that contains the information and documents referred to in subsections G.02.006(1) and (2).
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the senior person in charge of the site specified in the application; and
(b) include an attestation by that person that
(i) all of the information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the licensed dealer.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Renewal
G.02.011 (1) Subject to section G.02.013, on completion of the review of the renewal application, the Minister must issue a renewed dealer’s licence that contains the information specified in section G.02.007.
Marginal note:Terms and conditions
(2) When renewing a dealer’s licence, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to it or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence or the new level required as a result of the licence renewal; or
(c) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
- SOR/2004-238, s. 5
- SOR/2010-222, s. 7
- SOR/2019-171, s. 1
G.02.011.1 [Repealed, SOR/2019-171, s. 1]
G.02.011.2 [Repealed, SOR/2019-171, s. 1]
Marginal note:Validity
G.02.012 A renewed dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section G.02.027 or G.02.028.
- SOR/2004-238, s. 5
- SOR/2019-171, s. 1
Marginal note:Refusal
G.02.013 (1) The Minister must refuse to renew a dealer’s licence if
(a) the licensed dealer may no longer apply for a licence under section G.02.002;
(b) during the 10 years before the day on which the renewal application is submitted, the licensed dealer has contravened
(i) a provision of the Act, the Cannabis Act or their Regulations, or
(ii) a term or condition of a licence or permit issued to the dealer under a regulation made under the Act or issued to the dealer under the Cannabis Act or its regulations;
(c) during the 10 years before the day on which the renewal application is submitted, the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subparagraph G.02.005(a)(i) or (b)(i) or received a sentence as specified in subparagraph G.02.005(a)(ii) or (b)(ii);
(d) an activity for which the renewal is requested would contravene an international obligation;
(e) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the renewal is requested;
(f) the method referred to in paragraph G.02.006(1)(j) does not permit the recording of information as required by section G.02.071;
(g) the licensed dealer has not complied with the requirements of subsection G.02.010(3) or the information or documents that they have provided are not sufficient to complete the review of the renewal application;
(h) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the renewal application;
(i) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a controlled drug to an illicit market or use or has been involved in an activity that contravened an international obligation; or
(j) the Minister has reasonable grounds to believe that the renewal of the licence would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to renew a licence under paragraph (1)(b) or (h) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to renew a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Amendment of Licence
Marginal note:Application
G.02.014 (1) Before making a change affecting any information referred to in section G.02.007 that is contained in their dealer’s licence, a licensed dealer must submit to the Minister an application to amend the licence that contains a description of the proposed amendment, as well as the information and documents referred to in section G.02.006 that are relevant to the proposed amendment.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the senior person in charge of the site specified in the application; and
(b) include an attestation by that person that
(i) all of the information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the licensed dealer.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
- SOR/78-427, s. 1
- SOR/97-228, s. 10
- SOR/2004-238, s. 6
- SOR/2010-222, s. 8(E)
- SOR/2019-171, s. 1
Marginal note:Amendment
G.02.015 (1) Subject to section G.02.017, on completion of the review of the amendment application, the Minister must amend the dealer’s licence.
Marginal note:Terms and conditions
(2) When amending a dealer’s licence, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to it or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence or the new level required as a result of the amendment; or
(c) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
- SOR/78-427, s. 2
- SOR/2004-238, s. 7
- SOR/2010-222, s. 9
- SOR/2019-171, s. 1
Marginal note:Validity
G.02.016 An amended dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section G.02.027 or G.02.028.
Marginal note:Refusal
G.02.017 (1) The Minister must refuse to amend a dealer’s licence if
(a) an activity for which the licence amendment is requested would contravene an international obligation;
(b) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the licence amendment is requested;
(c) the method referred to in paragraph G.02.006(1)(j) does not permit the recording of information as required by section G.02.071;
(d) the licensed dealer has not complied with the requirements of subsection G.02.014(3) or the information or documents that they have provided are not sufficient to complete the review of the amendment application;
(e) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the amendment application; or
(f) the Minister has reasonable grounds to believe that the amendment of the licence would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to amend a licence under paragraph (1)(e) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to amend a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Changes Requiring Prior Approval by Minister
Marginal note:Application
G.02.018 (1) A licensed dealer must obtain the Minister’s approval before making any of the following changes by submitting a written request to the Minister:
(a) a change affecting the security measures at the site specified in the dealer’s licence;
(b) the replacement of the senior person in charge;
(c) the replacement of the qualified person in charge; or
(d) the replacement or addition of an alternate qualified person in charge.
Marginal note:Information and documents
(2) The licensed dealer must provide the Minister with the following with respect to a change referred to in subsection (1):
(a) in the case of a change affecting the security measures in place at the site specified in the dealer’s licence;
(b) in the case of the senior person in charge,
(i) the information specified in paragraph G.02.006(1)(c), and
(ii) the declaration specified in paragraph G.02.006(2)(b) and the documents specified in paragraphs G.02.006(2)(c) and (d); and
(c) in the case of the qualified person in charge or an alternate qualified person in charge,
(i) the information specified in paragraph G.02.006(1)(d), and
(ii) the declarations specified in paragraphs G.02.006(2)(b) and (e) and the documents specified in paragraphs G.02.006(2)(c), (d) and (f).
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
- SOR/2004-238, s. 8
- SOR/2010-222, s. 10
- SOR/2019-171, s. 1
Marginal note:Approval
G.02.019 (1) Subject to section G.02.020, on completion of the review of the application for approval of the change, the Minister must approve the change.
Marginal note:Terms and conditions
(2) When approving a change, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to the licence or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence; or
(c) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
- SOR/88-482, s. 2(F)
- SOR/2019-171, s. 1
Marginal note:Refusal
G.02.020 (1) The Minister must refuse to approve the change if
(a) during the 10 years before the day on which the application for approval of the change is submitted, the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subparagraph G.02.005(a)(i) or (b)(i) or received a sentence as specified in subparagraph G.02.005(a)(ii) or (b)(ii);
(b) the licensed dealer has not complied with the requirements of subsection G.02.018(3) or the information or documents that they have provided are not sufficient to complete the review of the application for approval of the change;
(c) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the application for approval of the change; or
(d) the Minister has reasonable grounds to believe that the change would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Exception
(2) The Minister must not refuse to approve a change under paragraph (1)(c) if the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use.
Marginal note:Notice
(3) Before refusing to approve a change, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard in respect of them.
Changes Requiring Notice to Minister
Marginal note:Prior notice
G.02.021 (1) A licensed dealer must notify the Minister in writing before
(a) making or assembling a product or compound that is not set out in the most recent version of the list referred to in paragraph G.02.006(1)(f) that has been submitted to the Minister; or
(b) making a change to a product or compound that is set out in the list, if the change affects any of the information that has previously been submitted.
Marginal note:Information and list
(2) The notice must contain the information referred to in paragraph G.02.006(1)(f) that is necessary to update the list and be accompanied by the revised version of the list.
Marginal note:Notice — five days
G.02.022 A licensed dealer must notify the Minister in writing within five days after a person ceases to act as the qualified person in charge or an alternate qualified person in charge.
Marginal note:Notice — 10 days
G.02.023 (1) A licensed dealer must notify the Minister in writing within 10 days after one of the following changes occurs:
(a) a person ceases to act as the senior person in charge; or
(b) the licensed dealer ceases to manufacture or assemble a product or compound that is set out in the most recent version of the list referred to in paragraph G.02.006(1)(f) has been submitted to the Minister.
Marginal note:Information and list
(2) A notice submitted under paragraph (1)(b) must specify which information referred to in paragraph G.02.006(1)(f) is being changed and be accompanied by the revised version of the list.
Marginal note:Notice of cessation of activities
G.02.024 (1) A licensed dealer that intends to cease conducting activities at their site — whether on or before the expiry of their licence — must notify the Minister in writing to that effect at least 30 days before ceasing those activities.
Marginal note:Content of notice
(2) The notice must be signed and dated by the senior person in charge and contain the following information:
(a) the expected date of the cessation of activities at the site;
(b) a description of the manner in which any remaining controlled drugs on the site as of that date will be disposed of by the licensed dealer, including
(i) if some or all of them will be sold or provided to another licensed dealer that will be conducting activities at the same site, the name of that dealer,
(ii) if some or all of them will be sold or provided to another licensed dealer that will not be conducting activities at the same site, the name of that dealer and the municipal address of their site, and
(iii) if some or all of them will be destroyed, the date on which and the municipal address of the location at which the destruction is to take place;
(c) the municipal address of the location at which the licensed dealer’s documents will be kept after activities have ceased; and
(d) the name, municipal address, telephone number and, if applicable, the email address of a person who the Minister may contact for further information after activities have ceased.
Marginal note:Update
(3) After having ceased to conduct the activities, the licensed dealer must submit to the Minister a detailed update of the information referred to in subsection (2) if it differs from what was set out in the notice. The update must be signed and dated by the senior person in charge.
- SOR/78-220, s. 3
- SOR/85-550, s. 2
- SOR/99-125, s. 2
- SOR/2004-238, s. 9
- SOR/2010-222, s. 11
- SOR/2012-230, s. 9
- SOR/2019-171, s. 1
G.02.024.1 [Repealed, SOR/2019-171, s. 1]
G.02.024.2 [Repealed, SOR/2019-171, s. 1]
Changes to Terms and Conditions of Licence
Marginal note:Addition of or modification to term or condition
G.02.025 (1) The Minister may, at any time other than the issuance, renewal or amendment of a dealer’s licence, add a term or condition to it or modify one if the Minister has reasonable grounds to believe that it is necessary to do so to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence; or
(c) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before adding a term or condition to a licence or modifying one, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Urgent circumstances
(3) Despite subsection (2), the Minister may add a term or condition to a licence or modify one without prior notice if the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use.
Marginal note:Urgent circumstances — notice
(4) The addition or modification of a term or condition that is made under subsection (3) takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the addition or modification;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
- SOR/78-220, s. 4
- SOR/78-427, s. 4
- SOR/85-550, s. 3
- SOR/88-482, s. 3(F)
- SOR/90-261, s. 2(F)
- SOR/97-228, s. 11
- SOR/2004-238, s. 11
- SOR/2010-222, s. 12
- SOR/2012-230, s. 10
- SOR/2014-260, ss. 6(E), 14(F)
- SOR/2019-171, s. 1
Marginal note:Deletion of term or condition
G.02.026 (1) The Minister may delete a term or condition of a dealer’s licence that the Minister determines is no longer necessary.
Marginal note:Notice
(2) The deletion takes effect as soon as the Minister sends the licensed dealer a notice to that effect.
- SOR/2004-238, s. 12
- SOR/2010-222, s. 13(F)
- SOR/2014-260, s. 7
- SOR/2019-171, s. 1
Suspension and Revocation of Licence
Marginal note:Suspension
G.02.027 (1) The Minister must suspend a dealer’s licence without prior notice if the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of licence
(3) The Minister must reinstate the licence if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
Marginal note:Revocation
G.02.028 (1) Subject to subsection (2), the Minister must revoke a dealer’s licence if
(a) the licensed dealer may no longer apply for a licence under section G.02.002;
(b) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the licence or the actual or potential unauthorized use of the licence;
(c) the licensed dealer ceases to conduct activities at their site before the expiry of their licence;
(d) the licensed dealer does not take the corrective measures specified in an undertaking or notice;
(e) the licensed dealer has contravened
(i) a provision of the Act, the Cannabis Act or their regulations, or
(ii) a term or condition of a licence or permit issued to the dealer under a regulation made under the Act or issued to the dealer under the Cannabis Act or its regulations;
(f) during the 10 years before the day on which the licence is revoked, the senior person in charge, the qualified person in charge or any alternate qualified person in charge was convicted as specified in subparagraph G.02.005(a)(i) or (b)(i) or received a sentence as specified in subparagraph G.02.005(a)(ii) or (b)(ii);
(g) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of an application relating to the licence; or
(h) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a controlled drug to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not revoke a dealer’s licence for a ground set out in paragraph (1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Return of licence
G.02.029 The licensed dealer must return the original of the licence to the Minister within 15 days after the effective date of the revocation.
Import Permits
Marginal note:Application
G.02.030 (1) A licensed dealer must submit to the Minister, before each importation of a controlled drug, an application for an import permit that contains the following information:
(a) their name, municipal address and dealer’s licence number;
(b) with respect to the controlled drug to be imported,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt,
(iii) its quantity, and
(iv) in the case of a raw material, its purity and its anhydrous content;
(c) if the controlled drug is contained in a product to be imported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the controlled drug in the product, the number of units per package and the number of packages;
(d) the name and municipal address, in the country of export, of the exporter from whom the controlled drug is being obtained;
(e) the name of the customs office where the importation is anticipated; and
(f) each proposed mode of transportation and any proposed country of transit or transhipment.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that all of the information submitted in support of the application is correct and complete to the best of their knowledge.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
G.02.031 Subject to section G.02.034, on completion of the review of the import permit application, the Minister must issue to the licensed dealer an import permit that contains
(a) the permit number;
(b) the information set out in subsection G.02.030(1);
(c) the effective date of the permit;
(d) the expiry date of the permit, being the earlier of
(i) a date specified by the Minister that is not more than 180 days after its effective date, and
(ii) the expiry date of the dealer’s licence; and
(e) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected, or
(ii) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Validity
G.02.032 An import permit is valid until the earliest of
(a) the expiry date set out in the permit,
(b) the date of the suspension or revocation of the permit under section G.02.037 or G.02.038,
(c) the date of the suspension or revocation of the dealer’s licence under section G.02.027 or G.02.028, and
(d) the date of the suspension or revocation of the export permit that applies to the controlled drug to be imported and that is issued by the competent authority in the country of export.
Marginal note:Return of permit
G.02.033 If an import permit expires, the licensed dealer must return the original of the permit to the Minister within 15 days after its expiry.
Marginal note:Refusal
G.02.034 (1) The Minister must refuse to issue an import permit if
(a) the licensed dealer is not authorized by their dealer’s licence to import the relevant controlled drug or their licence will expire before the date of importation;
(b) the Minister has reasonable grounds to believe that the importation would contravene an international obligation;
(c) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of the importation;
(d) the licensed dealer has not complied with the requirements of subsection G.02.030(3) or the information or documents that they have provided are not sufficient to complete the review of the permit application;
(e) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the permit application;
(f) the licensed dealer has been notified that their application to renew or amend their licence will be refused;
(g) the Minister has reasonable grounds to believe that the importation would contravene the laws of the country of export or any country of transit or transhipment; or
(h) the Minister has reasonable grounds to believe that the issuance of the permit would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before refusing to issue the import permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Providing copy of permit
G.02.035 The holder of an import permit must provide a copy of the permit to the customs office at the time of importation.
Marginal note:Declaration
G.02.036 The holder of an import permit must provide the Minister, within 15 days after the day of release of the controlled drug specified in the permit in accordance with the Customs Act, with a declaration that contains the following information:
(a) their name and the numbers of their dealer’s licence and the import permit that applies to the controlled drug;
(b) with respect to the controlled drug,
(i) its name, as set out in the dealer’s licence,
(ii) if it is a salt, the name of the salt, and
(iii) its quantity;
(c) if the controlled drug is contained in a product that they have imported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the controlled drug in the product, the number of units per package and the number of packages; and
(d) the name of the customs office from which the controlled drug was released and the date of the release.
Marginal note:Suspension
G.02.037 (1) The Minister must suspend an import permit without prior notice if
(a) the dealer’s licence is suspended;
(b) the Minister has reasonable grounds to believe that the suspension is necessary to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use; or
(c) the importation would contravene the laws of the country of export or any country of transit or transhipment.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of permit
(3) The Minister must reinstate the import permit if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
Marginal note:Revocation
G.02.038 (1) Subject to subsection (2), the Minister must revoke an import permit if
(a) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the permit or the actual or potential unauthorized use of the permit;
(b) the licensed dealer does not carry out the corrective measures specified by the Minister under paragraph G.02.037(2)(c) by the specified date;
(c) the licensed dealer has contravened a term or condition of the permit;
(d) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of the application for the permit;
(e) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a controlled drug to an illicit market or use; or
(f) the dealer’s licence has been revoked.
Marginal note:Exceptions
(2) The Minister must not revoke an import permit for a ground set out in paragraph (1)(d) or G.02.028(1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking an import permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Return of permit
G.02.039 If an import permit is revoked, the licensed dealer must return the original of the permit to the Minister within 15 days after the effective date of the revocation.
Export Permits
Marginal note:Application
G.02.040 (1) A licensed dealer must submit to the Minister, before each exportation of a controlled drug, an application for an export permit that contains the following information and document:
(a) their name, municipal address and dealer’s licence number;
(b) with respect to the controlled drug to be exported,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt,
(iii) its quantity, and
(iv) in the case of a raw material, its purity and its anhydrous content;
(c) if the controlled drug is contained in a product to be exported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the controlled drug in the product, the number of units per package and the number of packages;
(d) the name and municipal address of the importer in the country of final destination;
(e) the name of the customs office where the exportation is anticipated;
(f) each proposed mode of transportation and any proposed country of transit or transhipment; and
(g) a copy of the import permit issued by the competent authority in the country of final destination that sets out the name of the importer and the municipal address of their site in that country.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that, to the best of their knowledge,
(i) the exportation does not contravene the laws of the country of final destination or any country of transit or transhipment, and
(ii) all of the information and documents submitted in support of the application are correct and complete.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
G.02.041 Subject to section G.02.044, on completion of the review of the export permit application, the Minister must issue to the licensed dealer an export permit that contains
(a) the permit number;
(b) the information set out in paragraphs G.02.040(1)(a) to (f);
(c) the effective date of the permit;
(d) the expiry date of the permit, being the earliest of
(i) a date specified by the Minister that is not more than 180 days after its effective date,
(ii) the expiry date of the dealer’s licence, and
(iii) the expiry date of the import permit issued by the competent authority in the country of final destination; and
(e) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected, or
(ii) reduce a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Validity
G.02.042 An export permit is valid until the earliest of
(a) the expiry date set out in the permit,
(b) the date of the suspension or revocation of the permit under section G.02.047 or G.02.048,
(c) the date of the suspension or revocation of the dealer’s licence under section G.02.027 or G.02.028, and
(d) the date of the expiry, suspension or revocation of the import permit that applies to the controlled drug to be exported and that is issued by the competent authority in the country of final destination.
Marginal note:Return of permit
G.02.043 If an export permit expires, the licensed dealer must return the original of the permit to the Minister within 15 days after its expiry.
Marginal note:Refusal
G.02.044 (1) The Minister must refuse to issue an export permit if
(a) the licensed dealer is not authorized by their dealer’s licence to export the relevant controlled drug or their dealer’s licence will expire before the date of export;
(b) the Minister has reasonable grounds to believe that the exportation would contravene an international obligation;
(c) the licensed dealer has not complied with the requirements of subsection G.02.040(3) or the information or documents that they have provided are not sufficient to complete the review of the permit application;
(d) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the permit application;
(e) the licensed dealer has been notified that their application to renew or amend their licence will be refused;
(f) the Minister has reasonable grounds to believe that the exportation would not be in conformity with the import permit issued by the competent authority of the country of final destination;
(g) the Minister has reasonable grounds to believe that the exportation would contravene the laws of the country of final destination or any country of transit or transhipment; or
(h) the Minister has reasonable grounds to believe that the issuance of the permit would likely create a risk to public health or safety, including the risk of a controlled drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before refusing to issue the export permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Providing copy of permit
G.02.045 The holder of an export permit must provide a copy of the permit to the customs office at the time of exportation.
Marginal note:Declaration
G.02.046 The holder of an export permit must provide the Minister, within 15 days after the day of export of the controlled drug specified in the permit, with a declaration that contains the following information:
(a) their name and the numbers of their dealer’s licence and the export permit that applies to the controlled drug;
(b) with respect to the controlled drug,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt, and
(iii) its quantity;
(c) if the controlled drug is contained in a product that they have exported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the controlled drug in the product, the number of units per package and the number of packages; and
(d) the name of the customs office from which the controlled drug was exported and the date of export.
Marginal note:Suspension
G.02.047 (1) The Minister must suspend an export permit without prior notice if
(a) the dealer’s licence is suspended;
(b) the Minister has reasonable grounds to believe that the suspension is necessary to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use; or
(c) the exportation would contravene the laws of the country of final destination or any country of transit or transhipment.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of permit
(3) The Minister must reinstate the export permit if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
Marginal note:Revocation
G.02.048 (1) Subject to subsection (2), the Minister must revoke an export permit if
(a) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the permit or the actual or potential unauthorized use of the permit;
(b) the licensed dealer does not carry out the corrective measures specified by the Minister under paragraph G.02.047(2)(c) by the specified date;
(c) the licensed dealer has contravened a term or condition of the permit;
(d) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of the application for the permit;
(e) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a controlled drug to an illicit market or use; or
(f) the dealer’s licence has been revoked.
Marginal note:Exceptions
(2) The Minister must not revoke an export permit for a ground set out in paragraph (1)(d) or G.02.028(1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a controlled drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking an export permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Return of permit
G.02.049 If an export permit is revoked, the licensed dealer must return the original of the permit to the Minister within 15 days after the effective date of the revocation.
Identification
Marginal note:Name
G.02.050 A licensed dealer must include their name, as set out in their dealer’s licence, on all the means by which they identify themself in regard to their activities in relation to controlled drugs, including labels, orders, shipping documents, invoices and advertising
Sale of Controlled Drugs
Marginal note:Sale to another licensed dealer
G.02.051 A licensed dealer may sell or provide a controlled drug to another licensed dealer.
Marginal note:Sale to pharmacist
G.02.052 (1) Subject to subsection (2), a licensed dealer may sell or provide a controlled drug to a pharmacist.
Marginal note:Exception — pharmacist named in notice
(2) A licensed dealer must not sell or provide to a pharmacist named in a notice issued under section G.03.017.2 the controlled drugs referred to in the notice unless the dealer has received a notice of retraction issued under section G.03.017.3.
Marginal note:Sale to practitioner
G.02.053 (1) Subject to subsection (2), a licensed dealer may sell or provide a controlled drug to a practitioner.
Marginal note:Exception — practitioner named in notice
(2) A licensed dealer must not sell or provide to a practitioner who is named in a notice issued under section G.04.004.2 the controlled drugs referred to in the notice, unless the dealer has received a notice of retraction issued under section G.04.004.3.
Marginal note:Provision to hospital employee
G.02.054 A licensed dealer may provide a controlled drug to a hospital employee.
Marginal note:Sale to exempted person
G.02.055 A licensed dealer may sell or provide a controlled drug to a person who is exempted under section 56 of the Act with respect to the possession of that drug.
Marginal note:Sale to Minister
G.02.056 A licensed dealer may sell or provide a drug to the Minister.
Marginal note:Written order
G.02.057 A licensed dealer may sell or provide a controlled drug under any of sections G.02.051 to G.02.055 if
(a) the dealer has received a written order that specifies the name and quantity of the drug to be supplied and is signed and dated
(i) in the case of a drug to be provided to a hospital employee or a practitioner in a hospital, by the pharmacist in charge of the hospital’s pharmacy or by a practitioner authorized by the person in charge of the hospital to sign the order, and
(ii) in any other case, by the person to whom the drug is to be sold or provided; and
(b) the dealer has verified the signature, if it is unknown to them.
Marginal note:Verbal order
G.02.058 (1) A licensed dealer may sell or provide a controlled drug listed in Part II or III of the schedule to this Part if
(a) the dealer has received a verbal order that specifies the name and quantity of the drug to be supplied; and
(b) in the case of the provision of the drug to a hospital employee or a practitioner in a hospital, the order has been placed by the pharmacist in charge of the hospital’s pharmacy or by a practitioner authorized by the person in charge of the hospital to place the order.
Marginal note:Receipt
(2) A licensed dealer that has received a verbal order from a pharmacist or practitioner must, within five working days after filling the order, obtain and keep a receipt that includes
(a) the signature of the pharmacist or practitioner who received the controlled drug;
(b) the date on which the pharmacist or practitioner received the controlled drug; and
(c) the name and quantity of the controlled drug.
Marginal note:No further sale without receipt
(3) If the licensed dealer has not obtained the receipt within five working days, the dealer must not sell or provide a controlled drug to the pharmacist or practitioner in accordance with a further verbal order received from them until after obtaining the receipt.
Marginal note:Anticipated multiple sales
G.02.059 (1) A licensed dealer may sell or provide a controlled drug more than once in respect of one order if the order indicates
(a) the number of sales or provisions, not exceeding four;
(b) the specific quantity for each sale or provision; and
(c) the intervals between each sale or provision.
Marginal note:Multiple sales — insufficient stock
(2) A licensed dealer may sell or provide a controlled drug more than once in respect of one order if, at the time of receipt of the order, the dealer temporarily does not have in stock the quantity of the drug ordered, in which case the dealer may sell or provide against the order the quantity of the drug that the dealer has available and deliver the balance later.
Packaging and Transportation
Marginal note:Packaging — sale and provision
G.02.060 (1) A licensed dealer that sells or provides a controlled drug must securely package it in its immediate container, which must be sealed in such a manner that the container cannot be opened without breaking the seal.
Marginal note:Packaging — transport and export
(2) A licensed dealer that transports or exports a controlled drug must ensure that its package is sealed in such a manner that the package cannot be opened without breaking the seal.
Marginal note:Exception
(3) Subsection (1) does not apply to a test kit that contains a controlled drug and that has a registration number.
Marginal note:Transport
G.02.061 (1) A licensed dealer must, in taking delivery of a controlled drug that they have imported or in making delivery of a controlled drug,
(a) take any measures that are necessary to ensure the security of the drug while it is being transported;
(b) subject to subsection (2), use a method of transportation that permits an accurate record to be kept of all handling of the drug as well as the signatures of every person handling it until it is delivered to the consignee;
(c) in the case of an imported drug, transport it directly to the site specified in their licence after it is released under the Customs Act; and
(d) in the case of a drug to be exported, transport it directly from the site specified in their licence to the customs office where it will be exported.
Marginal note:Exception
(2) A licensed dealer may have a preparation transported by a common carrier.
Thefts, Losses and Suspicious Transactions
Marginal note:Protective measures
G.02.062 A licensed dealer must take any measures that are necessary to ensure the security of any controlled drug in their possession and any licence or permit in their possession.
Marginal note:Theft or loss — licence and permit
G.02.063 A licensed dealer that becomes aware of a theft or loss of their licence or permit must provide a written report to the Minister within 72 hours after becoming aware of it.
Marginal note:Theft or unexplainable loss — controlled drug
G.02.064 A licensed dealer that becomes aware of a theft of a controlled drug or of a loss of a controlled drug that cannot be explained on the basis of normally accepted business activities must
(a) provide a written report to a member of a police force within 24 hours after becoming aware of the theft or loss; and
(b) provide a written report to the Minister within 72 hours after becoming aware of the theft or loss and include a confirmation that the report required under paragraph (a) has been provided.
Marginal note:Suspicious transaction
G.02.065 (1) A licensed dealer must provide a written report containing the following information to the Minister within 72 hours after becoming aware of a transaction occurring in the course of their activities that they have reasonable grounds to suspect may be related to the diversion of a controlled drug to an illicit market or use:
(a) their name, municipal address, telephone number and, if the licensed dealer is a corporation, the position held by the individual making the report;
(b) the name and municipal address of the other party to the transaction;
(c) details of the transaction, including its date and time, its type, the name and quantity of the controlled drug and, in the case of a product or compound, the quantity of every controlled drug that it contains;
(d) in the case of a product that contains the controlled drug, other than a test kit, the drug identification number that is assigned to the product under section C.01.014.2; and
(e) a detailed description of the reasons for those suspicions.
Marginal note:Good faith
(2) No civil proceedings lie against a licensed dealer for having provided the report in good faith.
Marginal note:Non-disclosure
(3) A licensed dealer must not disclose that they have provided the report or disclose details of it, with the intent to prejudice a criminal investigation, whether or not a criminal investigation has begun.
Marginal note:Partial protection against self-incrimination
G.02.066 A report made under any of sections G.02.063 to G.02.065, or any evidence derived from it, is not to be used or received to incriminate the licensed dealer in any criminal proceeding against them other than a prosecution under section 132, 136 or 137 of the Criminal Code.
Destruction of Controlled Drugs
Marginal note:Destruction at site
G.02.067 A licensed dealer that intends to destroy a controlled drug at the site specified in their licence must ensure that the following conditions are met:
(a) the licensed dealer obtains the prior approval of the Minister;
(b) the destruction occurs in the presence of two of the following persons, at least one of whom must be a person referred to in subparagraph (i):
(i) the senior person in charge, the qualified person in charge or an alternate qualified person in charge, and
(ii) a person who works for or provides services to the licensed dealer and holds a senior position;
(c) the destruction is carried out in accordance with a method that complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction; and
(d) as soon as the destruction is completed, the person who carried out the destruction and each of the two persons referred to in paragraph (b) who were present at the destruction sign and date a joint declaration attesting that the controlled drug was completely destroyed, to which each signatory must add their name in printed letters.
Marginal note:Destruction elsewhere than at site
G.02.068 A licensed dealer that intends to destroy a controlled drug elsewhere than at the site specified in their licence must ensure that the following conditions are met:
(a) the licensed dealer obtains the prior approval of the Minister;
(b) the licensed dealer takes any measures that are necessary to ensure the security of the controlled drug while it is being transported in order to prevent its diversion to an illicit market or use;
(c) the destruction is carried out by a person working for a business that specializes in the destruction of dangerous goods and in the presence of another person working for that business;
(d) the destruction is carried out in accordance with a method that complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction; and
(e) as soon as the destruction is completed, the person who carried out the destruction provides the licensed dealer with a dated declaration attesting that the controlled drug was completely destroyed and containing
(i) the municipal address of the place of destruction,
(ii) the name and quantity of the controlled drug and, if applicable, the brand name and quantity of the product containing it or the name and quantity of the compound containing it,
(iii) the method of destruction,
(iv) the date of destruction, and
(v) the names in printed letters and signatures of that person and the other person who was present at the destruction.
Marginal note:Application for prior approval
G.02.069 (1) A licensed dealer must submit to the Minister an application that contains the following information in order to obtain the Minister’s prior approval to destroy a controlled drug:
(a) their name, municipal address and dealer’s licence number;
(b) the proposed date of destruction;
(c) the municipal address of the place of destruction;
(d) a brief description of the method of destruction;
(e) if the destruction is to be carried out at the site specified in the dealer’s licence, the names of the persons proposed for the purpose of paragraph G.02.067(b) and information establishing that they meet the conditions of that paragraph;
(f) the name of the controlled drug and, if applicable, the brand name of the product containing it or the name of the compound containing it; and
(g) the form and quantity of the controlled drug or the product or compound containing it and, if applicable, the strength per unit of the controlled drug in the product or compound, the number of units per package and the number of packages.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that
(i) the proposed method of destruction complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction, and
(ii) all of the information submitted in support of the application is correct and complete to the best of the signatory’s knowledge.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Approval
G.02.070 On completion of the review of the approval application, the Minister must approve the destruction of the controlled drug unless
(a) in the case of a destruction that is to be carried out at the site specified in the dealer’s licence, the persons proposed for the purpose of paragraph G.02.067(b) do not meet the conditions of that paragraph;
(b) the Minister has reasonable grounds to believe that the controlled drug would not be destroyed;
(c) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the approval application;
(d) the controlled drug or a portion of it is required for the purposes of a criminal or administrative investigation or a preliminary inquiry, trial or other proceeding under any Act or its regulations; or
(e) the Minister has reasonable grounds to believe that the approval would likely create a risk to public health or safety, including the risk of the controlled drug being diverted to an illicit market or use.
Documents
Marginal note:Method of recording information
G.02.071 A licensed dealer must record any information that they are required to record under this Part using a method that permits an audit of it to be made at any time.
Marginal note:Information — general
G.02.072 A licensed dealer must record the following information:
(a) the name, form and quantity of any controlled drug that the dealer orders, the name of the person who placed the order on the dealer’s behalf and the date of the order;
(b) the name, form and quantity of any controlled drug that the dealer receives, the name and municipal address of the person who sold or provided it and the date on which it was received;
(c) in the case of a controlled drug that the dealer sells or provides,
(i) the brand name of the product or the name of the compound containing the controlled drug and the name of the controlled drug,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any,
(iii) the form and quantity of the controlled drug and, if applicable, the strength per unit of the controlled drug in the product or compound, the number of units per package and the number of packages,
(iv) the name and municipal address of the person to whom it was sold or provided, and
(v) the date on which it was sold or provided;
(d) the name, form and quantity of any controlled drug that the dealer manufactures or assembles and the date on which it was placed in stock and, if applicable, the strength per unit of the controlled drug in the product or compound, the number of units per package and the number of packages;
(e) the name and quantity of any controlled drug that the dealer uses in the manufacturing or assembling of a product or compound, as well as the brand name and quantity of the product or the name and quantity of the compound, and the date on which the product or compound was placed in stock;
(f) the name, form and quantity of any controlled drug in stock at the end of each month;
(g) the name, form and quantity of any controlled drug that the dealer delivers, transports or sends, the name and municipal address of the consignee and the date on which it was delivered, transported or sent;
(h) the name, form and quantity of any controlled drug that the dealer imports, the date on which it was imported, the name and municipal address of the exporter, the country of exportation and any country of transit or transhipment; and
(i) the name, form and quantity of any controlled drug that the dealer exports, the date on which it was exported, the name and municipal address of the importer, the country of final destination and any country of transit or transhipment.
Marginal note:Verbal order
G.02.073 A licensed dealer that receives a verbal order for a controlled drug listed in Part II or III of the schedule to this Part and sells or provides it to a pharmacist, practitioner or hospital employee must immediately record
(a) the name of the person who placed the order;
(b) the date on which the order was received; and
(c) the name of the person recording the order.
Marginal note:Explainable loss of controlled drug
G.02.074 A licensed dealer that becomes aware of a loss of a controlled drug that can be explained on the basis of normally accepted business activities must record the following information:
(a) the name of the lost controlled drug and, if applicable, the brand name of the product or the name of the compound containing it;
(b) the form and quantity of the controlled drug and, if applicable, the form of the product or compound containing it, the strength per unit of the controlled drug in the product or compound, the number of units per package and the number of packages;
(c) the date on which the dealer became aware of the loss; and
(d) the explanation for the loss.
Marginal note:Destruction
G.02.075 A licensed dealer must record the following information concerning any controlled drug that they destroy at the site specified in their licence:
(a) the municipal address of the place of destruction;
(b) the name, form and quantity of the controlled drug and, if applicable, the brand name and quantity of the product containing the drug or the name and quantity of the compound containing the drug;
(c) the method of destruction; and
(d) the date of destruction.
Marginal note:Annual report
G.02.076 (1) Subject to subsections (2) and (3), a licensed dealer must provide to the Minister, within three months after the end of each calendar year, an annual report that contains
(a) the name, form and total quantity of each controlled drug that they receive, produce, sell, provide, import, export or destroy during the calendar year, as well as the name and total quantity of each controlled drug that they use to manufacture or assemble a product or compound;
(b) the name, form and quantity of each controlled drug in physical inventory taken at the site specified in their licence at the end of the calendar year; and
(c) the name, form and quantity of any controlled drug that has been lost or stolen in the course of conducting activities during the calendar year.
Marginal note:Non-renewal or revocation within first three months
(2) If a licensed dealer’s licence expires without being renewed or is revoked during the first three months of a calendar year, the dealer must provide to the Minister
(a) within three months after the end of the preceding calendar year, the annual report in respect of that year; and
(b) within three months after the expiry or revocation, a report in respect of the portion of the current calendar year during which the licence was valid that contains the information referred to in subsection (1), in which the quantity in physical inventory is to be calculated as of the date of expiry or revocation.
Marginal note:Non-renewal or revocation after third month
(3) If a licensed dealer’s licence expires without being renewed or is revoked after the first three months of a calendar year, the dealer must provide to the Minister, within three months after the expiry or revocation, a report in respect of the portion of the calendar year during which the licence was valid that contains the information referred to in subsection (1) for that period, in which the quantity in physical inventory is to be calculated as of the date of expiry or revocation.
Marginal note:Retention period
G.02.077 A licensed dealer and a former licensed dealer must keep any document containing the information that they are required to record under this Part, including every declaration and a copy of every report, for a period of two years following the day on which the last record is recorded in the document and in a manner that permits an audit of the document to be made at any time.
Marginal note:Location
G.02.078 The documents must be kept
(a) in the case of a licensed dealer, at the site specified in their licence; and
(b) in the case of a former licensed dealer, at a location in Canada.
Marginal note:Quality of documents
G.02.079 The documents must be complete and readily retrievable and the information in them must be legible and indelible.
DIVISION 3Pharmacists
- SOR/2019-171, s. 2(F)
Record of Controlled Drugs Received
- SOR/2019-171, s. 2
Marginal note:General information
G.03.001 (1) A pharmacist, on receipt of a controlled drug from a licensed dealer or from another pharmacist, shall keep a record of the name and quantity of the controlled drug received by them, the name and address of the person who sold or provided it and the date it was received.
Marginal note:Record
(2) The record of information referred to in subsection (1) shall be kept
(a) in a manner that permits an audit to be made; and
(b) subject to subsection (3), in a book, register or similar record maintained exclusively for controlled drugs.
Marginal note:Exception
(3) The record of information referred to in subsection (1) may, with respect to a controlled drug listed in Part II or III of the schedule to this Part, be kept in a form other than that specified in paragraph (2)(b).
- SOR/78-427, s. 5
- SOR/85-550, s. 4
- SOR/86-91, s. 2(F)
- SOR/90-261, s. 3(F)
- SOR/97-228, s. 12
- SOR/2004-238, s. 13
- SOR/2010-222, s. 14(E)
Sale of Controlled Drugs
- SOR/2019-171, s. 3
Marginal note:No sale without prescription
G.03.002 No pharmacist shall, except as otherwise provided in this Part, sell or provide a controlled drug to any person unless the pharmacist has first been provided with a prescription for it, and
(a) if the prescription is in writing, it has been signed and dated by the practitioner issuing the same and the signature of the practitioner where not known to the pharmacist, has been verified by him; or
(b) if the prescription is given verbally, the pharmacist has taken reasonable precaution to satisfy himself that the person giving the prescription is a practitioner.
- SOR/2004-238, s. 14
Marginal note:Prohibition — pharmacist or practitioner named in notice
G.03.002.1 Subject to section G.03.002.2 and notwithstanding sections G.03.002, G.03.003 and G.03.005, no pharmacist shall
(a) sell or provide a controlled drug, other than a preparation, to a pharmacist named in a notice given by the Minister under section G.03.017.2;
(b) sell or provide a preparation to a pharmacist named in a notice given by the Minister under section G.03.017.2;
(c) dispense, sell or provide a controlled drug, other than a preparation, to, or pursuant to a prescription or order given by, a practitioner named in a notice given by the Minister under section G.04.004.2; or
(d) dispense, sell or provide a preparation to a practitioner or pursuant to a prescription or order given by a practitioner named in a notice given by the Minister under section G.04.004.2.
- SOR/2003-135, ss. 7, 8
- SOR/2004-238, s. 15
- SOR/2019-171, s. 4(F)
Marginal note:Exception — notice of retraction
G.03.002.2 Section G.03.002.1 does not apply to a pharmacist to whom the Minister has issued a notice of retraction of the notice
(a) under section G.03.017.3, in respect of a pharmacist named in a notice issued by the Minister under section G.03.017.2; or
(b) under section G.04.004.3, in respect of a practitioner named in a notice issued by the Minister under section G.04.004.2.
- SOR/2003-135, s. 4
Marginal note:Sale to practitioner – order
G.03.003 (1) A pharmacist may sell or provide a controlled drug to a practitioner for use in their practice
(a) upon a written order, signed and dated by that practitioner, that has been verified if the signature of the practitioner is unknown to the pharmacist; or
(b) upon a verbal order specifying the name and quantity of the drug if the pharmacist has taken reasonable precautions to satisfy themself that the person making the order is a practitioner.
(2) [Repealed, SOR/2019-171, s. 5]
- SOR/85-550, s. 5
- SOR/2004-238, s. 16
- SOR/2012-230, s. 11
- SOR/2019-171, s. 5
Marginal note:Prescription file
G.03.004 A pharmacist shall, in respect of controlled drugs sold or provided to a practitioner under section G.03.003, keep in a special prescription file a record showing the date, the name and address of the practitioner, and the quantity and kind of controlled drug sold or provided.
- SOR/2004-238, s. 17
Marginal note:Provision to hospital
G.03.005 A pharmacist may provide a controlled drug to a hospital employee or to a practitioner in a hospital on receipt of a written order signed and dated by the pharmacist in charge of the hospital’s pharmacy or by a practitioner authorized by the person in charge of the hospital to sign the order, if the signature of that pharmacist or practitioner is known to the pharmacist or, if unknown, has been verified by the pharmacist.
- SOR/85-550, s. 6
- SOR/2004-238, s. 18(E)
- SOR/2019-171, s. 6
Marginal note:Refilling prescription
G.03.006 A pharmacist shall not refill a prescription for a controlled drug unless
(a) the practitioner, at the time that he issued the prescription, directed in writing, in the case of a controlled drug listed in Part I of the schedule to this Part, or directed in writing or orally, in the case of a controlled drug listed in Part II or III of the schedule to this Part, that the prescription be refilled, the number of times that it may be refilled and the dates for or the intervals between refills; and
(b) the pharmacist keeps a record of each refilling of a prescription.
- SOR/78-427, s. 6
- SOR/97-228, s. 13
Records
Marginal note:Written order or prescription
G.03.007 If, in accordance with a written order or prescription, a pharmacist dispenses a controlled drug listed in Part I of the schedule to this Part, other than a preparation, the pharmacist must immediately enter in a book, register or similar record maintained for such purposes
(a) their name or initials;
(b) the name, initials and municipal address of the practitioner who issued the order or prescription;
(c) the name and municipal address of the person named in the order or prescription;
(d) the name, form and quantity of the controlled drug dispensed;
(e) the date on which the controlled drug was dispensed; and
(f) the number assigned to the order or prescription.
- SOR/78-427, s. 7
- SOR/81-359, s. 1(F)
- SOR/97-228, s. 14
- SOR/2004-238
- s. 19
- SOR/2019-171, s. 7
Marginal note:Verbal order or prescription
G.03.008 A pharmacist must, before dispensing a controlled drug in accordance with a verbal order or prescription, make a written record of it that sets out
(a) their name or initials;
(b) the name, initials and municipal address of the practitioner who issued the order or prescription;
(c) the name and municipal address of the person named in the order or prescription;
(d) the name, form and quantity of the controlled drug;
(e) the directions for use given with the order or prescription;
(f) the date on which the controlled drug was dispensed; and
(g) the number assigned to the order or prescription.
- SOR/85-550, s. 7
- SOR/2004-238, s. 20
- SOR/2019-171, s. 7
Marginal note:File by date and number
G.03.009 A pharmacist must maintain a special prescription file in which are filed, in sequence as to date and number, all written orders and prescriptions for controlled drugs that they have dispensed and the written record of all controlled drugs that they have dispensed in accordance with a verbal order or prescription.
Marginal note:Retention period
G.03.010 A pharmacist shall retain in his possession for a period of at least two years, any records which he is required to keep by this Part.
General Obligations of Pharmacist
- SOR/2019-171, s. 8
Marginal note:Providing information and assisting inspector
G.03.011 A pharmacist shall
(a) furnish such information respecting the dealings of the pharmacist in any controlled drug in such form and at such times as the Minister may require;
(b) make available and produce to an inspector upon request his special prescription file together with any books, records or documents which he is required to keep;
(c) permit an inspector to make copies of or to take extracts from such files, books, records or documents; and
(d) permit an inspector to check all stocks of controlled drugs on his premises.
Marginal note:Loss or theft — protective measures
G.03.012 A pharmacist shall take all reasonable steps that are necessary to protect controlled drugs on his premises or under his control against loss or theft.
- SOR/85-550, s. 8
Marginal note:Loss or theft — report
G.03.013 A pharmacist shall report to the Minister any loss or theft of a controlled drug within 10 days of his discovery thereof.
Return, Sale and Transfer
- SOR/2019-171, s. 9
Marginal note:Written order
G.03.014 A pharmacist may, upon receiving a written order for a controlled drug signed and dated by
(a) the licensed dealer who sold or provided that drug to them, return that drug to that licensed dealer;
(b) another pharmacist, sell or provide any quantity of that drug to that other pharmacist that is specified in the order as being required for emergency purposes;
(c) the Minister, sell or provide to the Minister any quantity of that drug, specified in the order, that is required by the Minister in connection with his or her duties; and
(d) a person exempted under section 56 of the Act with respect to that controlled drug, sell or provide to that person any quantity of that drug that is specified in the order.
- SOR/81-359, s. 2
- SOR/85-550, s. 9
- SOR/99-125, s. 3
- SOR/2004-238, s. 21
- SOR/2014-260, s. 8(E)
- SOR/2018-69, s. 65
- SOR/2019-171, s. 10
Marginal note:Record
G.03.015 A pharmacist shall immediately after receiving, selling or providing a controlled drug under paragraph G.03.014(b) or (c) or subsection G.05.003(4) enter the details of the transaction in a book, register or other record maintained for the purpose of recording such transactions.
- SOR/85-550, s. 10
- SOR/2004-238, s. 22
Marginal note:Notice to Minister
G.03.016 A pharmacist shall forthwith after removing, transporting or transferring a controlled drug from his place of business to any other place of business operated by him notify the Minister, setting out the details.
Communication of Information by Minister to Licensing Authority
Marginal note:Contraventions by pharmacist
G.03.017 The Minister must provide in writing any factual information about a pharmacist that has been obtained under the Act or this Part to the provincial professional licensing authority that is responsible for the authorization of the person to practise their profession
(a) in the province in which the pharmacist is or was entitled to practise if
(i) the authority submits to the Minister a written request that sets out the pharmacist’s name and address, a description of the information being requested and a statement that the information is required for the purpose of assisting a lawful investigation by the authority, or
(ii) the Minister has reasonable grounds to believe that the pharmacist has
(A) contravened a rule of conduct established by the authority,
(B) been convicted of a designated substance offence, or
(C) contravened this Part; or
(b) in a province in which the pharmacist is not entitled to practise, if the authority submits to the Minister
(i) a written request that sets out the pharmacist’s name and address and a description of the information being requested, and
(ii) a document that shows that
(A) the pharmacist has applied to that authority to practise in that province, or
(B) the authority has reasonable grounds to believe that the pharmacist is practising in that province without being authorized to do so.
- SOR/86-881, s. 1
- SOR/97-228, s. 15
- SOR/2003-135, s. 5
- SOR/2010-222, s. 15
- SOR/2019-171, s. 11
Notice of Prohibition of Sale
Marginal note:Request by pharmacist
G.03.017.1 A pharmacist may make a written request to the Minister to send to the persons and authorities specified in subsection G.03.017.2(3) a notice, issued under section G.03.017.2, advising them that recipients of the notice must not sell or provide a controlled drug other than a preparation, a preparation, or both, to that pharmacist.
- SOR/2003-135, s. 5
Marginal note:Notice by Minister
G.03.017.2 (1) In the circumstances described in subsection (2), the Minister must send a notice to the persons and authorities specified in subsection (3) advising them that pharmacists practising in the notified pharmacies and licensed dealers must not sell or provide to the pharmacist named in the notice a controlled drug other than a preparation or a preparation.
Marginal note:Circumstances requiring a notice
(2) The notice must be sent if the pharmacist named in the notice has
(a) made a request to the Minister in accordance with section G.03.017.1 to send the notice;
(b) contravened a rule of conduct established by the provincial professional licensing authority of the province in which the pharmacist is practising and the authority has requested the Minister in writing to send the notice; or
(c) been convicted of a designated substance offence or of a contravention of this Part.
Marginal note:Recipients
(3) The notice must be sent to
(a) all licensed dealers;
(b) all pharmacies within the province in which the pharmacist named in the notice is entitled to practice and is practising;
(c) the provincial professional licensing authority of the province in which the pharmacist named in the notice is entitled to practise;
(d) all pharmacies in an adjacent province in which an order from the pharmacist named in the notice may be filled; and
(e) any provincial professional licensing authority in another province that has requested the Minister in writing to send the notice.
Marginal note:Other circumstances
(4) The Minister may send the notice described in subsection (1) to the persons and authorities specified in subsection (3) if the Minister has taken the measures specified in subsection (5) and has reasonable grounds to believe that the pharmacist named in the notice
(a) has contravened a provision of the Act or this Part;
(b) has, on more than one occasion, self-administered a controlled drug, other than a preparation, contrary to accepted pharmaceutical practice;
(c) has, on more than one occasion, self-administered a preparation, contrary to accepted pharmaceutical practice;
(d) has, on more than one occasion, provided or administered a controlled drug, other than a preparation, to a person who is a spouse, common-law partner, parent or child of the pharmacist, including a child adopted in fact, contrary to accepted pharmaceutical practice;
(e) has, on more than one occasion, provided or administered a preparation to a person who is a spouse, common-law partner, parent or child of the pharmacist, including a child adopted in fact, contrary to accepted pharmaceutical practice; or
(f) is unable to account for the quantity of controlled drug for which the pharmacist was responsible under this Part.
Marginal note:Measures before sending notice
(5) The measures that must be taken before sending the notice are that the Minister has
(a) consulted with the provincial professional licensing authority of the province in which the pharmacist to whom the notice relates is entitled to practise;
(b) given that pharmacist an opportunity to be heard; and
(c) considered
(i) the compliance history of the pharmacist in respect of the Act and its regulations, and
(ii) whether the actions of the pharmacist pose a risk to public health or safety, including the risk of the controlled drug being diverted to an illicit market or use.
- SOR/2003-135, s. 5
- SOR/2010-222, ss. 16, 35(F)
- SOR/2019-171, s. 12
Marginal note:Notice of retraction
G.03.017.3 The Minister must provide the licensed dealers, pharmacies and provincial professional licensing authorities who were sent a notice under subsection G.03.017.2(1) with a notice of retraction of that notice if
(a) in the circumstance described in paragraph G.03.017.2(2)(a), the requirements set out in subparagraphs (b)(i) and (ii) have been met and one year has elapsed since the notice was sent by the Minister; or
(b) in a circumstance described in any of paragraphs G.03.017.2(2)(b) and (c) and (4)(a) to (f), the pharmacist named in the notice has
(i) requested in writing that a retraction of the notice be sent, and
(ii) provided a letter from the provincial professional licensing authority of the province in which the pharmacist is entitled to practise, in which the authority consents to the retraction of the notice.
- SOR/2003-135, s. 5
- SOR/2010-222, s. 17
- SOR/2019-171, s. 12
G.03.017.4 and G.03.017.5 [Repealed, SOR/2003-135, s. 5]
DIVISION 4Practitioners
Administration of Designated Drugs and Other Controlled Drugs
Marginal note:Restriction
G.04.001 (1) Subject to subsections (2) and (3) and to an exemption granted under section 56 of the Act with respect to the administration of the controlled drug specified in the exemption, a practitioner must not administer a controlled drug to any person or animal.
Marginal note:Conditions
(2) A practitioner may administer a controlled drug, other than a designated drug, to a person or to an animal, if
(a) that person or animal is under their professional treatment; and
(b) the controlled drug is required for the condition for which that person or animal is receiving treatment.
Marginal note:Purposes
(3) A practitioner of medicine, dentistry or veterinary medicine or a nurse practitioner may administer a designated drug to a person or animal who is under their professional treatment if the designated drug is for the treatment of any of the following conditions:
(a) in the case of persons,
(i) narcolepsy,
(ii) hyperkinetic disorders in children,
(iii) epilepsy,
(iv) parkinsonism, or
(v) hypotensive states associated with anesthesia; or
(b) in the case of animals, depression of cardiac and respiratory centres.
Marginal note:Definitions
(4) The following definitions apply in this section.
- administer
administer includes to prescribe, sell or provide a drug. (administrer)
- designated drug
designated drug means any of the following controlled drugs:
(a) amphetamine and its salts;
(b) benzphetamine and its salts;
(c) methamphetamine and its salts;
(d) phenmetrazine and its salts; or
(e) phendimetrazine and its salts. (drogue désignée)
- SOR/99-125, s. 4
- SOR/2004-238, s. 23
- SOR/2012-230, s. 12
- SOR/2019-171, s. 13
Records
Marginal note:Record of controlled drugs sold or provided
G.04.002 (1) A practitioner who sells or provides a controlled drug to a person for self-administration or for administration to an animal shall, whether or not the practitioner charges for the drug, keep a record showing the name and quantity of the controlled drug sold or provided, the name and address of the person to whom it was sold or provided and the date on which it was sold or provided if the quantity of the controlled drug exceeds
(a) three times the maximum daily dosage recommended by the manufacturer or assembler of the controlled drug; or
(b) three times the generally recognized maximum daily therapeutic dosage for that controlled drug if the manufacturer or assembler has not recommended a maximum daily dosage.
Marginal note:Accessibility of record
(2) A practitioner who is required by this section to keep a record shall keep the record in a place, form and manner that will permit an inspector readily to examine and obtain information from it.
- SOR/88-482, s. 4(F)
- SOR/2004-238, s. 24
- SOR/2019-171, s. 14
General Obligations of Practitioner
- SOR/2019-171, s. 15
Marginal note:Requirements
G.04.002A A practitioner shall
(a) furnish to the Minister on request such information respecting
(i) the use by the practitioner of controlled drugs received — including the administering, selling or providing of the drugs to a person — , and
(ii) the prescriptions for controlled drugs issued by the practitioner,
as the Minister may require;
(b) produce to an inspector on request any records that these Regulations require the practitioner to keep;
(c) permit an inspector to make copies of such records or to take extracts therefrom;
(d) permit an inspector to check all stocks of controlled drugs on the practitioner’s premises;
(e) retain in his possession for at least two years any record that these Regulations require him to keep;
(f) take adequate steps to protect controlled drugs in his possession from loss or theft; and
(g) report to the Minister any loss or theft of a controlled drug within 10 days of the practitioner’s discovery of the loss or theft.
- SOR/2004-238, s. 25
G.04.003 [Repealed, SOR/2010-222, s. 18]
Communication of Information by Minister to Licensing Authority
Marginal note:Contraventions by practitioner
G.04.004 The Minister must provide in writing any factual information about a practitioner that has been obtained under the Act or this Part to the provincial professional licensing authority responsible for the registration and authorization of the person to practise their profession
(a) in the province in which the practitioner is or was registered and entitled to practise if
(i) the authority submits to the Minister a written request that sets out the practitioner’s name and address, a description of the information being requested and a statement that the information is required for the purpose of assisting a lawful investigation by the authority, or
(ii) the Minister has reasonable grounds to believe that the practitioner has
(A) contravened a rule of conduct established by the authority,
(B) been convicted of a designated substance offence, or
(C) contravened this Part; or
(b) in a province in which the practitioner is not registered and entitled to practise, if the authority submits to the Minister
(i) a written request that sets out the practitioner’s name and address and a description of the information being requested, and
(ii) a document that shows that
(A) the practitioner has applied to that authority to practise in that province, or
(B) the authority has reasonable grounds to believe that the practitioner is practising in that province without being authorized to do so.
- SOR/86-881, s. 2
- SOR/97-228, s. 17
- SOR/2003-135, s. 6
- SOR/2010-222, s. 19
- SOR/2019-171, s. 16
Notice of Prohibition of Sale
Marginal note:Request by practitioner
G.04.004.1 A practitioner may make a written request to the Minister to send to licensed dealers and pharmacies a notice, issued under section G.04.004.2, advising them of one or more of the following requirements:
(a) recipients of the notice must not sell or provide a controlled drug, other than a preparation, to that practitioner;
(b) recipients of the notice must not sell or provide a preparation to that practitioner;
(c) pharmacists practising in the notified pharmacies must not fill a prescription or order for a controlled drug, other than a preparation, from that practitioner; and
(d) pharmacists practising in the notified pharmacies must not fill a prescription or order for a preparation from that practitioner.
- SOR/2003-135, s. 6
Marginal note:Notice by Minister
G.04.004.2 (1) In the circumstances described in subsection (2), the Minister must send a notice to the persons and authorities specified in subsection (3) advising them that
(a) pharmacists practising in the notified pharmacies and licensed dealers must not sell or provide to the practitioner named in the notice a controlled drug other than a preparation or a preparation;
(b) pharmacists practising in the notified pharmacies must not fill a prescription or order from the practitioner named in the notice for a controlled drug other than a preparation or a preparation; or
(c) the prohibitions in both paragraphs (a) and (b) apply with respect to the practitioner named in the notice.
Marginal note:Circumstances requiring a notice
(2) The notice must be sent if the practitioner named in the notice has
(a) made a request to the Minister in accordance with section G.04.004.1 to send the notice;
(b) contravened a rule of conduct established by the provincial professional licensing authority of the province in which the practitioner is practising and the authority has requested the Minister in writing to issue the notice; or
(c) been convicted of a designated substance offence or of a contravention of this Part.
Marginal note:Recipients
(3) The notice must be sent to
(a) all licensed dealers;
(b) all pharmacies within the province in which the practitioner named in the notice is registeredand entitled to practice and is practising;
(c) the provincial professional licensing authority of the province in which the practitioner named in the notice is registered and entitled to practise;
(d) all pharmacies in an adjacent province in which a prescription or order from the practitioner named in the notice may be filled; and
(e) any provincial professional licensing authority in another province that has requested the Minister in writing to send the notice; and
Marginal note:Other circumstances
(4) The Minister may send the notice described in subsection (1) to the persons and authorities specified in subsection (3) if the Minister has taken the measures specified in subsection (5) and has reasonable grounds to believe that the practitioner named in the notice
(a) has contravened a provision of the Act or this Part;
(b) has, on more than one occasion, self-administered a controlled drug, other than a preparation, under a self-directed prescription or order or, in the absence of a prescription or order, contrary to accepted professional practice;
(c) has, on more than one occasion, self-administered a preparation, under a self-directed prescription or order or, in the absence of a prescription or order, contrary to accepted professional practice;
(d) has, on more than one occasion, prescribed, provided or administered a controlled drug, other than a preparation, to a person who is a spouse, common-law partner, parent or child of the practitioner, including a child adopted in fact, contrary to accepted professional practice;
(e) has, on more than one occasion, prescribed, provided or administered a preparation to a person who is a spouse, common-law partner, parent or child of the practitioner, including a child adopted in fact, contrary to accepted professional; or
(f) is unable to account for the quantity of controlled drug for which the practitioner was responsible under this Part.
Marginal note:Measures before sending notice
(5) The measures that must be taken before sending the notice are that the Minister has
(a) consulted with the provincial professional licensing authority of the province in which the practitioner to whom the notice relates is registered and entitled to practise;
(b) given that practitioner an opportunity to be heard
(c) considered
(i) the compliance history of the practitioner in respect of the Act and its regulations, and
(ii) whether the actions of the practitioner pose a risk to public health or safety, including the risk of the controlled drug being diverted to an illicit market or use.
- SOR/2003-135, s. 6
- SOR/2010-222, ss. 20, 35(F)
- SOR/2019-171, s. 17
Marginal note:Notice of retraction
G.04.004.3 The Minister must provide the licensed dealers, pharmacies and provincial professional licensing authorities who were sent a notice under subsection G.04.004.2(1) with a notice of retraction of that notice if
(a) in the circumstance described in paragraph G.04.004.2(2)(a), the requirements set out in subparagraphs (b)(i) and (ii) have been met and one year has elapsed since the notice was sent by the Minister; or
(b) in a circumstance described in any of paragraphs G.04.004.2(2)(b) and (c) and (4)(a) to (f), the practitioner named in the notice has
(i) requested in writing that a retraction of the notice be sent, and
(ii) provided a letter from the provincial professional licensing authority of the province in which the practitioner is registered and entitled to practise, in which the authority consents to the retraction of the notice.
- SOR/88-482, s. 5(F)
- SOR/2003-135, s. 6
- SOR/2010-222, s. 21
- SOR/2019-171, s. 17
G.04.004.4 and G.04.004.5 [Repealed, SOR/2003-135, s. 6]
DIVISION 5Hospitals
Marginal note:Record — controlled drugs
G.05.001 (1) A person who is in charge of a hospital shall keep or cause to be kept a record of the following information:
(a) the name and quantity of any controlled drug received for the hospital by a hospital employee or a practitioner in the hospital;
(b) the name and address of the person from whom any controlled drug was received and the date on which it was received;
(c) the name and quantity of any controlled drug used in the making or assembling of a product or compound containing that controlled drug, the name and quantity of the product or compound made or assembled and the date on which the product or compound was placed in stock;
(c.1) the name and quantity of any controlled drug produced and the date on which it was placed in stock;
(d) the name of the patient for whom a controlled drug was dispensed;
(e) the name of the practitioner ordering or prescribing a controlled drug; and
(f) the date on which a controlled drug was ordered or prescribed and the form and quantity thereof.
Marginal note:Record keeping
(2) Subject to subsections (3) and (4), the record of information referred to in subsection (1) shall be kept
(a) in a manner that permits an audit to be made;
(b) in a book, register or similar record maintained exclusively for controlled drugs; and
(c) for a period of at least two years.
Marginal note:Exception — preparation
(3) The information referred to in paragraphs (1)(d) to (f) may, with respect to a preparation, be kept in a form other than that specified in paragraph (2)(b).
Marginal note:Exception — Part II or III of schedule
(4) The information referred to in subsection (1) may, with respect to a controlled drug listed in Part II or III of the schedule to this Part, be kept in a form other than that specified in paragraph (2)(b).
- SOR/78-427, s. 8
- SOR/85-550, s. 11
- SOR/88-482, s. 6
- SOR/97-228, s. 18
- SOR/2004-238, s. 27
- SOR/2019-171, s. 18(F)
Marginal note:Providing information and assisting inspector
G.05.002 A person who is in charge of a hospital shall
(a) furnish such information respecting the use of controlled drugs therein, in such form and at such times as the Minister may require;
(b) produce to an inspector any books, records or documents required by these Regulations to be kept;
(c) permit an inspector to make copies thereof or take extracts from such books, records and documents; and
(d) permit an inspector to check all stocks of controlled drugs in the hospital.
Marginal note:Selling, providing or administering controlled drug
G.05.003 (1) No person in charge of a hospital shall permit a controlled drug to be sold, provided or administered except in accordance with this section.
Marginal note:Prescription or written order
(2) On receipt of a prescription or a written order signed and dated by a practitioner, the person in charge of a hospital may permit a controlled drug to be administered to a person or an animal under treatment as an in-patient or out-patient of the hospital, or to be sold or provided to the person or to the person in charge of the animal.
Marginal note:Emergency — other hospital
(3) Subject to subsection (6), the person in charge of a hospital may permit a controlled drug to be provided, for emergency purposes, to a hospital employee or a practitioner in another hospital on receipt of a written order signed and dated by a pharmacist in the other hospital or a practitioner authorized by the person in charge of the other hospital to sign the order.
Marginal note:Emergency — pharmacist
(4) Subject to subsection (6), the person in charge of a hospital may permit a controlled drug to be sold or provided, for emergency purposes, to a pharmacist on receipt of a written order signed and dated by the pharmacist.
Marginal note:Research purposes
(5) The person in charge of a hospital may permit a controlled drug to be provided to a person employed in a research laboratory in that hospital for the purpose of research.
Marginal note:Signature
(6) No person in charge of a hospital shall permit a controlled drug to be sold or provided under subsection (3) or (4) unless the signature of the pharmacist in the other hospital or of the practitioner authorized by the person in charge of the other hospital to sign an order is known to the person who sells or provides the controlled drug or has been verified.
- SOR/85-550, s. 12
- SOR/88-482, s. 7
- SOR/2004-238, s. 28
- SOR/2010-222, s. 22(F)
- SOR/2019-171, s. 19(F)
- SOR/2019-171, s. 25(F)
Marginal note:Loss or theft
G.05.004 A person who is in charge of a hospital shall take all steps necessary to protect controlled drugs in the hospital against loss or theft and shall report to the Minister any loss or theft of a controlled drug within 10 days of his discovery thereof.
- SOR/78-427, s. 9
DIVISION 6General
Marginal note:Labelling — drug dispensed in accordance with prescription
G.06.001 In the case of a controlled drug that is dispensed by a pharmacist in accordance with a prescription, section C.01.004 does not apply but the label of the package in which the controlled drug is contained must include the following:
(a) the name and municipal address of the pharmacy or pharmacist;
(b) the date and number of the prescription;
(c) the name of the person for whom the controlled drug is dispensed;
(d) the name of the practitioner;
(e) directions for use; and
(f) any other information that the prescription requires be shown on the label.
- SOR/99-125, s. 5
- SOR/2004-238, s. 29
- SOR/2018-69, ss. 67, 69
- SOR/2019-171, s. 20
Marginal note:Labelling — test kit
G.06.002 Section C.01.004 does not apply to a test kit that contains a controlled drug and that has a registration number.
- SOR/85-550, s. 13
- SOR/88-482, s. 8(F)
- SOR/99-125, s. 6
- SOR/2019-171, s. 20
G.06.002.1 [Repealed, SOR/2019-171, s. 20]
G.06.002.2 [Repealed, SOR/2019-171, s. 20]
G.06.002.3 [Repealed, SOR/2019-171, s. 20]
G.06.002.4 [Repealed, SOR/2019-171, s. 20]
Marginal note:Identification or analysis of controlled drug
G.06.003 (1) Despite anything in this Part, a person may, for the purpose of identification or analysis of a controlled drug, provide or deliver the drug to
(a) a practitioner of medicine; or
(b) an agent or mandatary of a practitioner of medicine, if the agent or mandatary is exempted under section 56 of the Act with respect to the possession of that drug for that purpose.
Marginal note:Agent or mandatary of practitioner of medicine
(2) An agent or mandatary of a practitioner of medicine who receives the controlled drug must immediately provide or deliver it to
(a) the practitioner; or
(b) the Minister.
Marginal note:Practitioner of medicine
(3) A practitioner of medicine who receives the controlled drug must immediately provide or deliver it
(a) for the purpose of its identification or analysis, to a person exempted under section 56 of the Act with respect to the possession of that drug for that purpose; or
(b) to the Minister.
Marginal note:Advertising
G.06.004 It is prohibited to
(a) advertise a controlled drug to the general public; or
(b) publish any written advertisement respecting a controlled drug unless that advertisement displays the following symbol in a clear and conspicuous colour and size in the upper left quarter of its first page:

Marginal note:Record keeping — specific cases
G.06.005 Every person who is exempted under section 56 of the Act with respect to the possession or administration of a controlled drug and every practitioner of medicine who has received a controlled drug under subsection G.06.003(1) or (2) and every agent or mandatary of a practitioner of medicine who has received a controlled drug under subsection G.06.003(1) must
(a) keep a record of the following information for a two-year period beginning on the day on which the record is made:
(i) the name and quantity of any controlled drug purchased or received by them and the date on which it was purchased or received,
(ii) the name and address of the person from whom the controlled drug was purchased or received, and
(iii) details of the use of the controlled drug;
(b) provide any information respecting those controlled drugs that the Minister may require; and
(c) permit access to the records required to be kept by this Part.
Marginal note:Communication of information by Minister to nursing statutory body
G.06.006 (1) The Minister may provide to a nursing statutory body any information concerning any member of that body that has been obtained under this Part, the Act or the Food and Drugs Act.
Marginal note:Non-application
(2) Subsection (1) does not apply to a nurse practitioner.
Marginal note:Definitions
(3) The following definitions apply in this section.
- member
member means any person who is authorized by a nursing statutory body to practice nursing. (membre)
- nursing statutory body
nursing statutory body means any provincial professional licensing authority that, in accordance with the laws of that province, authorizes a person to practise nursing. (organisme régissant la profession d’infirmier)
Marginal note:Notification of application for order of restoration
G.06.007 (1) For the purpose of subsection 24(1) of the Act, notification of an application for an order of restoration must be given in writing to the Attorney General by registered mail and be mailed not less than 15 days before the date on which the application is to be made to a justice.
Marginal note:Content of notification
(2) The notification must specify
(a) the name of the justice to whom the application is to be made;
(b) the time and place at which the application is to be heard;
(c) details concerning the controlled drug or other thing in respect of which the application is to be made; and
(d) the evidence on which the applicant intends to rely to establish that they are entitled to possession of the controlled drug or other thing referred to in paragraph (c).
G.07.001 [Repealed, SOR/2019-171, s. 20]
G.07.002 [Repealed, SOR/2019-171, s. 20]
SCHEDULE(Sections G.01.001 and G.01.002, subsections G.01.005(1) and G.02.058(1), section G.02.073, subsection G.03.001(3), paragraph G.03.006(a), section G.03.007 and subsection G.05.001(4))
PART I
- 1Amphetamines, their salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues, excluding those substances set out in item 1 of Part I of the schedule to Part J but including:
- (1)amphetamine (α-methylbenzeneethanamine)
- (2)methamphetamine (N,α-dimethylbenzeneethanamine)
- (3)Benzphetamine (N-benzyl-N,α-dimethylbenzeneethanamine)
- 2Methylphenidate (methyl 2-phenyl-2-(piperidin-2-yl)acetate), its salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues, including:
- (1)Ethylphenidate (ethyl 2-phenyl-2-(piperidin-2-yl)acetate)
- (2)Isopropylphenidate (isopropyl 2-phenyl-2-(piperidin-2-yl)acetate)
- (3)Propylphenidate (propyl 2-phenyl-2-(piperidin-2-yl)acetate)
- (4)3,4-Dichloromethylphenidate (methyl 2-(3,4-dichlorophenyl)-2-(piperidin-2-yl)acetate)
- (5)4-Methylmethylphenidate (methyl 2-(4-methylphenyl)-2-(piperidin-2-yl)acetate)
- (6)4-Fluoromethylphenidate (methyl 2-(4-fluorophenyl)-2-(piperidin-2-yl)acetate)
- (7)Methylnaphthidate (methyl 2-(naphthalen-2-yl)-2-(piperidin-2-yl)acetate)
- (8)Ethylnaphthidate (ethyl 2-(naphthalen-2-yl)-2-(piperidin-2-yl)acetate)
- 3Methaqualone (2-methyl-3-(2-methylphenyl)-4(3H)quinazolinone) and any salt thereof
- 4Phendimetrazine (d-3,4-dimethyl-2-phenylmorpholine) and any salt thereof
- 5Phenmetrazine (3-methyl-2-phenylmorpholine) and any salt thereof
- 6Pentobarbital (5-ethyl-5-(1-methylbutyl)barbituric acid)
- 7Secobarbital (5-allyl-5-(1-methylbutyl)barbituric acid)
- 84-hydroxybutanoic acid (GHB) and any salt thereof
- 9Aminorex (5-phenyl-4,5-dihydro-1,3-oxazol-2-amine), its salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues, including
- (1)4-Methylaminorex (4-methyl-5-phenyl-4,5-dihydro-1,3-oxazol-2-amine)
- (2)4,4’-Dimethylaminorex (4-methyl-5-(4-methylphenyl)-4,5-dihydro-1,3-oxazol-2-amine)
- 10Fenetylline (d,l-3,7-dihydro-1,3-dimethyl-7-(2-[(1-methyl-2-phenethyl)amino]ethyl)-1H-purine-2,6-dione) and any salt thereof
- 11Glutethimide (2-ethyl-2-phenylglutarimide)
- 12Lefetamine ((-)-N,N-dimethyl-α-phenylbenzeneethanamine), its salts, derivatives and isomers and salts of derivatives and isomers
- 13Mecloqualone (2-methyl-3-(2-chlorophenyl)-4(3H)-quinazolinone) and any salt thereof
- 14Mesocarb (3-(α-methylphenethyl)-N-(phenylcarbamoyl)sydnone imine) and any salt thereof
- 15Pemoline (2-amino-5-phenyl-oxazolin-4-one) and any salt thereof
- 16Zipeprol (4-(2-methoxy-2-phenylethyl)-α-(methoxyphenylmethyl)-1-piperazineethanol) and any salt thereof
- 17Amineptine (7-[(10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)amino]heptanoic acid) and any salt thereof
PART II
- 1Barbiturates, their salts and derivatives, excluding the substances set out in items 6 and 7 of Part I of this schedule, as well as barbituric acid (2,4,6(1H,3H,5H)-pyrimidinetrione) and its salts and 1,3-dimethylbarbituric acid (1,3-dimethyl-2,4,6(1H,3H,5H)-pyrimidinetrione) and its salts, but including
- (1)Allobarbital (5,5-diallylbarbituric acid)
- (2)Alphenal (5-allyl-5-phenylbarbituric acid)
- (3)Amobarbital (5-ethyl-5-(3-methylbutyl)barbituric acid)
- (4)Aprobarbital (5-allyl-5-isopropylbarbituric acid)
- (5)Barbital (5,5-diethylbarbituric acid)
- (6)[Repealed, SOR/2017-12, s. 1]
- (7)Butabarbital (5-sec-butyl-5-ethylbarbituric acid)
- (8)Butalbital (5-allyl-5-isobutylbarbituric acid)
- (9)Butallylonal (5-(2-bromoallyl)-5-sec-butylbarbituric acid)
- (10)Butethal (5-butyl-5-ethylbarbituric acid)
- (11)Cyclobarbital (5-(1-cyclohexen-1-yl)-5-ethylbarbituric acid)
- (12)Cyclopal (5-allyl-5-(2-cyclopenten-1-yl)barbituric acid)
- (13)Heptabarbital (5-(1-cyclohepten-1-yl)-5-ethylbarbituric acid)
- (14)Hexethal (5-ethyl-5-hexylbarbituric acid)
- (15)Hexobarbital (5-(1-cyclohexen-1-yl)-1,5-dimethylbarbituric acid)
- (16)Mephobarbital (5-ethyl-1-methyl-5-phenylbarbituric acid)
- (17)Methabarbital (5,5-diethyl-1-methylbarbituric acid)
- (18)Methylphenobarbital (5-ethyl-1-methyl-5-phenylbarbituric acid)
- (19)Propallylonal (5-(2-bromoallyl)-5-isopropyl-barbituric acid)
- (20)Phenobarbital (5-ethyl-5-phenylbarbituric acid)
- (21)Probarbital (5-ethyl-5-isopropylbarbituric acid)
- (22)Phenylmethylbarbituric Acid (5-methyl-5-phenylbarbituric acid)
- (23)Sigmodal(5-(2-bromoallyl)-5-(1-methylbutyl)- barbituric acid)
- (24)Talbutal (5-allyl-5-sec-butylbarbituric acid)
- (25)Vinbarbital (5-ethyl-5-(1-methyl-1-butenyl)barbituric acid)
- (26)Vinylbital (5-(1-methylbutyl)-5-vinylbarbituric acid)
- 2Thiobarbiturates, their salts and derivatives, including:
- (1)Thialbarbital (5-allyl-5-(2-cyclohexen-1-yl)-2-thiobarbituric acid)
- (2)Thiamylal (5-allyl-5-(1-methylbutyl)-2-thiobarbituric acid)
- (3)Thiobarbituric Acid (2-thiobarbituric acid)
- (4)Thiopental(5-ethyl-5-(1-methylbutyl)-2- thiobarbituric acid)
- 3Chlorphentermine (1-(p-chlorophenyl)-2-methyl-2-aminopropane) and any salt thereof
- 4Diethylpropion (2-(diethylamino)propiophenone) and any salt thereof
- 5Phentermine (α,α-dimethylbenzeneethanamine) and any salt thereof
- 6Butorphanol (1-N-cyclobutylmethyl-3,14-dihydroxymorphinan) and its salts
- 7Nalbuphine (N-cyclobutylmethyl-4,5-epoxy-morphinan-3,6,14-triol) and any salt thereof
- 8Pyrovalerone (4′-methyl-2-(1-pyrrolidinyl)valerophenone) and any salt thereof
PART III
- 1Anabolic steroids and their derivatives, including:
- (1)Androisoxazole (17ß-hydroxy-17α-methylandrostano[3,2-c]isoxazole)
- (2)Androstanolone (17ß-hydroxy-5α-androstan-3-one)
- (3)Androstenediol (androst-5-ene-3ß,17ß-diol)
- (4)Bolandiol (estr-4-ene-3b,17ß-diol)
- (5)Bolasterone (17ß-hydroxy-7α,17-dimethylandrost-4-en-3-one)
- (6)Bolazine (17ß-hydroxy-2α-methyl-5α-androstan-3-one azine)
- (7)Boldenone (17ß-hydroxyandrosta-1,4-dien-3-one)
- (8)Bolenol (19-nor-17α-pregn-5-en-17-ol)
- (9)Calusterone (17ß-hydroxy-7b,17-dimethylandrost-4-en-3-one)
- (10)Clostebol (4-chloro-17ß-hydroxyandrost-4-en-3-one)
- (11)Drostanolone (17ß-hydroxy-2α-methyl-5α-androstan-3-one)
- (12)Enestebol (4,17ß-dihydroxy-17-methylandrosta-1,4-dien-3-one)
- (13)Epitiostanol (2α, 3α-epithio-5α-androstan-17ß-ol)
- (14)Ethylestrenol (19-nor-17α-pregn-4-en-17-ol)
- (15)4-Hydroxy-19-nor testosterone
- (16)Fluoxymesterone (9-fluoro-11ß,17ß-dihydroxy — 17-methylandrost-4-en-3-one)
- (17)Formebolone (11α,17ß-dihydroxy-17-methyl-3-oxoandrosta-1,4-dien-2-carboxaldehyde)
- (18)Furazabol (17-methyl-5α-androstano[2,3-c]furazan-17ß-ol)
- (19)Mebolazine (17ß-hydroxy-2α,17-dimethyl-5α-androstan-3-one azine)
- (20)Mesabolone (17ß-[(1-methoxycyclohexyl)oxy]-5α-androst-1-en-3-one)
- (21)Mesterolone (17ß-hydroxy-1α-methyl-5α-androstan-3-one)
- (22)Metandienone (17ß-hydroxy-17-methylandrosta-1,4-dien-3-one)
- (23)Metenolone (17ß-hydroxy-1-methyl-5α-androst-1-en-3-one)
- (24)Methandriol (17α-methylandrost-5-ene-3ß,17ß-diol)
- (25)Methyltestosterone (17ß-hydroxy-17-methyl-androst-4-en-3-one)
- (26)Metribolone (17ß-hydroxy-17-methylestra-4,9,11-trien-3-one)
- (27)Mibolerone (17ß-hydroxy-7α,17-dimethylestr-4-en-3-one)
- (28)Nandrolone (17ß-hydroxyestr-4-en-3-one)
- (29)Norboletone (13-ethyl-17ß-hydroxy-18,19-dinorpregn-4-en-3-one)
- (30)Norclostebol (4-chloro-17ß-hydroxyestr-4-en-3-one)
- (31)Norethandrolone (17α-ethyl-17ß-hydroxyestr-4-en-3-one)
- (32)Oxabolone (4,17ß-dihydroxyestr-4-en-3-one)
- (33)Oxandrolone (17ß-hydroxy-17-methyl-2-oxa-5α-androstan-3-one)
- (34)Oxymesterone (4,17ß-dihydroxy-17-methylandrost-4-en-3-one)
- (35)Oxymetholone (17ß-hydroxy-2-(hydroxymethylene)-17-methyl-5α-androstan-3-one)
- (36)Prasterone (3ß-hydroxyandrost-5-en-17-one)
- (37)Quinbolone (17ß-(1-cyclopenten-1-yloxy)androsta-1,4-dien-3-one)
- (38)Stanozolol (17ß-hydroxy-17-methyl-5α-androstano[3,2-c]pyrazole)
- (39)Stenbolone (17ß-hydroxy-2-methyl-5α-androst-1-en-3-one)
- (40)Testosterone (17ß-hydroxyandrost-4-en-3-one)
- (41)Tibolone ((7α,17α)-17-hydroxy-7-methyl-19-norpregn-5(10)en-20-yn-3-one)
- (42)Tiomesterone (1α,7α-bis(acetylthio)-17ß-hydroxy-17-methylandrost-4-en-3-one)
- (43)Trenbolone (17ß-hydroxyestra-4,9,11-trien-3-one)
- 2Zeranol (3,4,5,6,7,8,9,10,11,12-decahydro-7,14,16-trihydroxy-3-methyl-1H-2-benzoxacyclotetradecin-1-one)
- SOR/78-427, s. 10
- SOR/79-753, s. 1
- SOR/81-84, s. 1
- SOR/85-550, s. 14(F)
- SOR/86-678, s. 1
- SOR/89-381, s. 1
- SOR/92-386, s. 3
- SOR/97-228, s. 21
- SOR/99-425, s. 1
- SOR/2003-34, ss. 2, 3
- SOR/2003-413, s. 2
- SOR/2015-210, s. 1
- SOR/2016-106, s. 1
- SOR/2017-12, ss. 1, 2(E)
- SOR/2017-43, s. 1
- SOR/2017-250, s. 1
- SOR/2019-171, s. 21
PART JRestricted Drugs
Definitions
Marginal note:Definitions
J.01.001 The following definitions apply in this Part.
- Act
Act means the Controlled Drugs and Substances Act. (Loi)
- competent authority
competent authority means a public authority of a foreign country that is authorized under the laws of the country to approve the importation or exportation of restricted drugs into or from the country. (autorité compétente)
- compound
compound includes a preparation. (composé)
- designated criminal offence
designated criminal offence means
(a) an offence involving the financing of terrorism against any of sections 83.02 to 83.04 of the Criminal Code;
(b) an offence involving fraud against any of sections 380 to 382 of the Criminal Code;
(c) the offence of laundering proceeds of crime against section 462.31 of the Criminal Code;
(d) an offence involving a criminal organization against any of sections 467.11 to 467.13 of the Criminal Code; or
(e) a conspiracy or an attempt to commit, being an accessory after the fact in relation to, or any counselling in relation to, an offence referred to in any of paragraphs (a) to (d). (infraction désignée en matière criminelle)
- destroy
destroy, in respect of a restricted drug, means to alter or denature it to such an extent that its consumption is rendered impossible or improbable. (destruction)
- hospital
hospital means a facility that is
(a) licensed, approved or designated by a province in accordance with the laws of the province to provide care or treatment to persons or animals suffering from any form of disease or illness; or
(b) owned or operated by the Government of Canada or the government of a province and that provides health services. (hôpital)
- institution
institution means any institution engaged in research on drugs and includes a hospital, a university in Canada or a department or agency of the Government of Canada or of a government of a province or any part of them. (établissement)
- international obligation
international obligation means an obligation in respect of a restricted drug set out in a convention, treaty or other multilateral or bilateral instrument that Canada has ratified or to which Canada adheres. (obligation internationale)
- label
label has the same meaning as in section 2 of the Food and Drugs Act. (étiquette)
- licensed dealer
licensed dealer means the holder of a licence issued under section J.01.015. (distributeur autorisé)
- package
package includes anything in which a restricted drug is wholly or partly contained, placed or packed. (emballage)
- pharmacist
pharmacist[Repealed, SOR/2021-271, s. 1]
- prescription
prescription[Repealed, SOR/2021-271, s. 1]
- proper name
proper name, in respect of a restricted drug, means the name in English or French that
(a) is assigned to the drug in section C.01.002;
(b) appears in bold face type for the drug in these Regulations and, if the drug is dispensed in a form other than that described in Part C, the name of the dispensing form; or
(c) is assigned in any of the publications mentioned in Schedule B to the Food and Drugs Act in the case of a drug not included in paragraph (a) or (b). (nom propre)
- qualified investigator
qualified investigator means, in respect of a restricted drug, a person whose use and possession of that drug are authorized by the Minister under subsection J.01.059(4) and who is
(a) employed by or connected with an institution; or
(b) engaged in clinical testing or laboratory research in an institution in respect of that drug. (chercheur compétent)
- qualified person in charge
qualified person in charge means the individual designated under subsection J.01.012(1). (responsable qualifié)
- restricted drug
restricted drug means a controlled substance that is set out in the schedule to this Part. (drogue d’usage restreint)
- Security Directive
Security Directive means the Directive on Physical Security Requirements for Controlled Substances and Drugs Containing Cannabis, as amended from time to time and published by the Government of Canada on its website. (Directive en matière de sécurité)
- senior person in charge
senior person in charge means the individual designated under section J.01.011. (responsable principal)
- test kit
test kit means a kit
(a) that contains a restricted drug and a reagent system or buffering agent;
(b) that is used during the course of a chemical or analytical procedure to test for the presence or quantity of a restricted drug for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose; and
(c) the contents of which are not intended or likely to be consumed by, or administered to, a person or an animal. (nécessaire d’essai)
- SOR/97-228, s. 22
- SOR/2004-238, s. 31
- SOR/2013-172, s. 1
- SOR/2019-171, s. 22
- SOR/2021-271, s. 1
General
Marginal note:Temporary accelerated scheduling
J.01.002 (1) The Minister may, by order, add to column 1 of Part III of the schedule to this Part any item or portion of an item listed in Schedule V to the Act for a period referred to in column 2 that is the same as that listed in Schedule V for that item.
Marginal note:Deletion
(2) The Minister may, by order, delete any item or portion of an item from column 1 of Part III of the schedule to this Part.
Marginal note:Deletion — Schedule V to Act
(3) An item or portion of an item listed in Part III of the schedule to this Part is deemed to be deleted on the day on which it is no longer listed in Schedule V to the Act.
- SOR/97-228, s. 23
- SOR/99-125, s. 7
- SOR/2010-222, s. 23
- SOR/2015-210, s. 2
- SOR/2018-85, s. 1
- SOR/2019-171, s. 22
Marginal note:Non-application — member of police force
J.01.003 A member of a police force or a person acting under their direction and control who, in respect of the conduct of the member or person, is exempt from the application of subsection 4(2) or section 5, 6 or 7 of the Act by virtue of the Controlled Drugs and Substances Act (Police Enforcement) Regulations is, in respect of that conduct, exempt from the application of this Part.
- SOR/2004-238, s. 32
- SOR/2019-171, s. 22
J.01.003.1 [Repealed, SOR/2019-171, s. 22]
J.01.003.2 [Repealed, SOR/2019-171, s. 22]
Possession
Marginal note:Authorized persons
J.01.004 (1) The following persons are authorized to possess a restricted drug listed in Part I of the schedule to this Part:
(a) a licensed dealer;
(b) a qualified investigator who possesses the drug for the purpose of conducting clinical testing or laboratory research in an institution;
(c) an inspector, member of the Royal Canadian Mounted Police, police constable, peace officer, member of the technical or scientific staff of the Government of Canada, the government of a province or a university in Canada who possesses the drug in connection with their employment;
(d) a person exempted under section 56 of the Act with respect to the possession of that drug; and
(e) the Minister.
Marginal note:Agent or mandatary
(2) A person is authorized to possess a restricted drug listed in Part I of the schedule to this Part if they are acting as the agent or mandatary of a person referred to in paragraph (1)(a), (b), (d) or (e).
Marginal note:Agent or mandatary — person referred to in paragraph (1)(c)
(3) A person is authorized to possess a restricted drug listed in Part I of the schedule to this Part if they
(a) are acting as the agent or mandatary of a person who they have reasonable grounds to believe is a person referred to in paragraph (1)(c); and
(b) possess the restricted drug for the purpose of assisting that person in the administration or enforcement of an Act or regulation.
J.01.004.1 [Repealed, SOR/2019-171, s. 22]
Test Kits
Marginal note:Authorized activities
J.01.005 A person may sell, possess or otherwise deal in a test kit if the following conditions are met:
(a) a registration number has been issued for the test kit under section J.01.007 and has not been cancelled under section J.01.008;
(b) the test kit bears, on its external surface,
(i) the name of the manufacturer,
(ii) the trade name or trademark, and
(iii) the registration number; and
(c) the test kit will be used for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose.
Marginal note:Application for registration number
J.01.006 (1) The manufacturer of a test kit may obtain a registration number for it by submitting to the Minister an application containing
(a) a detailed description of the design and construction of the test kit;
(b) a detailed description of the restricted drug and other substances, if any, contained in the test kit, including the qualitative and quantitative composition of each component; and
(c) a description of the proposed use of the test kit.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the person authorized by the applicant for that purpose; and
(b) include an attestation by that person that all of the information submitted in support of the application is correct and complete to the best of their knowledge.
Marginal note:Additional information or document
(3) The applicant must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance of registration number
J.01.007 On completion of the review of the application for a registration number, the Minister must issue a registration number for the test kit, preceded by the letters “TK”, if the Minister determines that the test kit will only be used for a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose and that it contains
(a) a restricted drug and an adulterating or denaturing agent that are combined in such a manner and in such a quantity, proportion or concentration that the preparation or mixture has no significant drug abuse potential; or
(b) such small quantities or concentrations of any restricted drug as to have no significant drug abuse potential.
- SOR/2004-238, s. 33
- SOR/2010-222, s. 25
- SOR/2014-260, ss. 9, 16(F)
- SOR/2019-171, s. 22
J.01.007.1 [Repealed, SOR/2019-171, s. 22]
J.01.007.2 [Repealed, SOR/2019-171, s. 22]
J.01.007.3 [Repealed, SOR/2019-171, s. 22]
J.01.007.4 [Repealed, SOR/2019-171, s. 22]
J.01.007.5 [Repealed, SOR/2019-171, s. 22]
J.01.007.6 [Repealed, SOR/2019-171, s. 22]
J.01.007.7 [Repealed, SOR/2019-171, s. 22]
J.01.007.8 [Repealed, SOR/2019-171, s. 22]
J.01.007.9 [Repealed, SOR/2019-171, s. 22]
J.01.007.91 [Repealed, SOR/2019-171, s. 22]
Marginal note:Cancellation of registration number
J.01.008 The Minister must cancel the registration number for a test kit if
(a) the test kit is removed from the market by the manufacturer;
(b) the Minister has reasonable grounds to believe that the test kit is used or is likely to be used for any purpose other than a medical, laboratory, industrial, educational, law administration or enforcement, or research purpose; or
(c) the Minister has reasonable grounds to believe that the cancellation is necessary to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use.
Licensed Dealers
Authorized Activities
Marginal note:General
J.01.009 (1) A licensed dealer may produce, assemble, sell, provide, transport, send, deliver, import or export a restricted drug if they comply with this Part and the terms and conditions of their dealer’s licence and any permit issued under this Part.
Marginal note:Qualified person in charge present
(2) A licensed dealer may conduct an activity in relation to a restricted drug at their site only if the qualified person in charge or an alternate qualified person in charge is present at the site.
Marginal note:Permit — import and export
(3) A licensed dealer must obtain a permit to import or export a restricted drug.
Marginal note:Possession for export
(4) A licensed dealer may possess a restricted drug for the purpose of exporting it if they have obtained it in accordance with this Part.
Dealer’s Licences
Preliminary Requirements
Marginal note:Eligible persons
J.01.010 The following persons may apply for a dealer’s licence:
(a) an individual who ordinarily resides in Canada;
(b) a corporation that has its head office in Canada or operates a branch office in Canada; or
(c) the holder of a position that includes responsibility for restricted drugs on behalf of the Government of Canada or of a government of a province, a police force, a hospital or a university in Canada.
Marginal note:Senior person in charge
J.01.011 An applicant for a dealer’s licence must designate only one individual as the senior person in charge, who has overall responsibility for management of the activities with respect to restricted drugs that are specified in the licence application. The applicant may designate themself if the applicant is an individual.
- SOR/2004-238, s. 34
- SOR/2019-171, s. 22
Marginal note:Qualified person in charge
J.01.012 (1) An applicant for a dealer’s licence must designate only one individual as the qualified person in charge, who is responsible for supervising the activities with respect to restricted drugs that are specified in the licence application and for ensuring that those activities comply with this Part. The applicant may designate themself if the applicant is an individual.
Marginal note:Alternate qualified person in charge
(2) An applicant for a dealer’s licence may designate an individual as an alternate qualified person in charge, who is authorized to replace the qualified person in charge when that person is absent. The applicant may designate themself if the applicant is an individual.
Marginal note:Qualifications
(3) Only an individual who meets the following requirements may be designated as a qualified person in charge or an alternate qualified person in charge:
(a) they work at the site specified in the dealer’s licence;
(b) they
(i) are a person entitled or, if applicable, registered and entitled by a provincial professional licensing authority or a professional association in Canada and entitled to practise a profession that is relevant to their duties, such as pharmacist, practitioner, pharmacy technician or laboratory technician,
(ii) hold a diploma, certificate or credential awarded by a post-secondary educational institution in Canada in a field or occupation that is relevant to their duties, such as pharmacy, medicine, dentistry, veterinary medicine, pharmacology, chemistry, biology, pharmacy technician, laboratory technician, pharmaceutical regulatory affairs or supply chain management or security, or
(iii) hold a diploma, certificate or credential that is awarded by a foreign educational institution in a field or occupation referred to in subparagraph (ii) and hold
(A) an equivalency assessment as defined in subsection 73(1) of the Immigration and Refugee Protection Regulations, or
(B) an equivalency assessment issued by an organization or institution that is responsible for issuing equivalency assessments and is recognized by a province;
(c) they have sufficient knowledge of and experience with the use and handling of the restricted drugs specified in the dealer’s licence to properly carry out their duties; and
(d) they have sufficient knowledge of the provisions of the Act and this Part that are applicable to the activities specified in the dealer’s licence to properly carry out their duties.
Marginal note:Exception
(4) An applicant for a dealer’s licence may designate an individual who does not meet any of the requirements of paragraph (3)(b) as a qualified person in charge or an alternate qualified person in charge if
(a) no other individual working at the site meets those requirements;
(b) those requirements are not necessary for the activities specified in the licence; and
(c) the individual has sufficient knowledge — acquired from a combination of education, training or work experience — to properly carry out their duties.
- SOR/2004-238, s. 34
- SOR/2010-222, s. 30
- SOR/2019-171, s. 22
J.01.012.1 [Repealed, SOR/2019-171, s. 22]
J.01.012.2 [Repealed, SOR/2019-171, s. 22]
Marginal note:Ineligibility
J.01.013 An individual is not eligible to be a senior person in charge, a qualified person in charge or an alternate qualified person in charge if, during the 10 years before the day on which the dealer’s licence application is submitted,
(a) in respect of a designated substance offence or a designated criminal offence or a designated offence as defined in subsection 2(1) of the Cannabis Act, the individual
(i) was convicted as an adult, or
(ii) was a young person who received an adult sentence, as those terms are defined in subsection 2(1) of the Youth Criminal Justice Act; or
(b) in respect of an offence committed outside Canada that, if committed in Canada, would have constituted a designated substance offence or a designated criminal offence or a designated offence as defined in subsection 2(1) of the Cannabis Act,
(i) the individual was convicted as an adult, or
(ii) if they committed the offence when they were at least 14 years old but less than 18 years old, the individual received a sentence that was longer than the maximum youth sentence, as that term is defined in subsection 2(1) of the Youth Criminal Justice Act, that could have been imposed under that Act for such an offence.
- SOR/2004-238, s. 34
- SOR/2019-171, s. 22
Issuance of Licence
Marginal note:Application
J.01.014 (1) A person who intends to conduct an activity referred to in section J.01.009 must obtain a dealer’s licence for each site at which they intend to conduct activities by submitting an application to the Minister that contains the following information:
(a) if the licence is requested by
(i) an individual, the individual’s name,
(ii) a corporation, its corporate name and any other name registered with a province under which it intends to conduct the activities specified in its dealer’s licence or by which it intends to identify itself, and
(iii) the holder of a position mentioned in paragraph J.01.010(c), the applicant’s name and the title of the position;
(b) the municipal address, telephone number and, if applicable, the email address of the proposed site and, if different from the municipal address, its mailing address;
(c) the name, date of birth, telephone number and email address of the proposed senior person in charge;
(d) with respect to each of the proposed qualified person in charge and any proposed alternate qualified person in charge,
(i) their name, date of birth, telephone number and email address,
(ii) the title of their position at the site,
(iii) the name and title of the position of their immediate supervisor at the site,
(iv) if applicable, the profession they practise that is relevant to their duties, the name of the province that authorizes them to practise it and their authorization number, and
(v) their education, training and work experience that are relevant to their duties;
(e) the activities that are to be conducted and the restricted drugs in respect of which each of the activities is to be conducted;
(f) if the licence is requested to manufacture or assemble a product or compound that contains a restricted drug, other than a test kit, a list that includes, for each product or compound,
(i) the brand name of the product or the name of the compound,
(ii) the name of the restricted drug in the product or compound,
(iii) the strength per unit of the restricted drug in it, the number of units per package and the number of packages,
(iv) if it is to be manufactured or assembled by or for another licensed dealer under a custom order, the name, municipal address and dealer’s licence number of the other licensed dealer, and
(v) if the applicant’s name appears on the label of the product or compound, a copy of the inner label;
(g) if the licence is requested in order to produce a restricted drug other than a product or compound that contains a restricted drug,
(i) the name of the restricted drug,
(ii) the quantity that the applicant expects to produce under their licence and the period during which that quantity would be produced, and
(iii) if it is to be produced for another licensed dealer under a custom order, the name, municipal address and licence number of the other licensed dealer;
(h) if the licence is requested for an activity that is not described in paragraph (f) or (g), the name of the restricted drug for which the activity is to be conducted and the purpose of the activity;
(i) a detailed description of the security measures in place at the site, determined in accordance with the Security Directive; and
(j) a detailed description of the method for recording information that the applicant proposes to use for the purpose of section J.01.075.
Marginal note:Documents
(2) An application for a dealer’s licence must be accompanied by the following documents:
(a) if the applicant is a corporation, a copy of
(i) the certificate of incorporation or other constituting instrument, and
(ii) any document filed with the province in which its site is located that states its corporate name and any other name registered with the province under which the applicant intends to conduct the activities specified in its dealer’s licence or by which it intends to identify itself;
(b) individual declarations signed and dated by each of the proposed senior person in charge, and qualified person in charge and any proposed alternate qualified person in charge, attesting that the person is not ineligible for a reason specified in section J.01.013;
(c) a document issued by a Canadian police force in relation to each person referred to in paragraph (b), indicating whether, during the 10 years before the day on which the application is submitted, the person was convicted as specified in subparagraph J.01.013(a)(i) or received a sentence as specified in subparagraph J.01.013(a)(ii);
(d) if any of the persons referred to in paragraph (b) has ordinarily resided in a country other than Canada during the 10 years before the day on which the application is submitted, a document issued by a police force of that country indicating whether in that period that person was convicted as specified in subparagraph J.01.013(b)(i) or received a sentence as specified in subparagraph J.01.013(b)(ii);
(e) declaration, signed and dated by the proposed senior person in charge, attesting that the proposed qualified person in charge and any proposed alternate qualified person in charge have the knowledge and experience required under paragraphs J.01.012(3)(c) and (d); and
(f) if the proposed qualified person in charge or any proposed alternate qualified person in charge does not meet the requirement of subparagraph J.01.012(3)(b)(i), either
(i) a copy of the person’s diploma, certificate or credential referred to in subparagraph J.01.012(3)(b)(ii) or (iii), or
(ii) a detailed description of the education, training and work experience that is required under paragraph J.01.012(4)(c), together with supporting evidence, such as a copy of a course transcript or an attestation by the person who provided the training.
Marginal note:Signature and attestation
(3) The application must
(a) be signed and dated by the proposed senior person in charge; and
(b) include an attestation by that person that
(i) all information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the applicant.
Marginal note:Additional information and documents
(4) The applicant must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
J.01.015 Subject to section J.01.017, on completion of the review of the licence application, the Minister must issue a dealer’s licence, with or without terms and conditions, that contains
(a) the licence number;
(b) the name of the licensed dealer, their corporate name or the title of the position they hold;
(c) the activities that are authorized and the names of the restricted drugs in respect of which each activity may be conducted;
(d) the municipal address of the site at which the dealer may conduct the authorized activities;
(e) the security level at the site, determined in accordance with the Security Directive;
(f) the effective date of the licence;
(g) the expiry date of the licence, which must be not later than three years after its effective date;
(h) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected,
(ii) ensure conformity with the requirements associated with the security level that is referred to in paragraph (e), or
(iii) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use; and
(i) if the licensed dealer produces a restricted drug, the quantity that they may produce and the authorized production period.
Marginal note:Validity
J.01.016 A dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section J.01.035 or J.01.036.
Marginal note:Refusal
J.01.017 (1) The Minister must refuse to issue a dealer’s licence if
(a) the applicant may not apply for a licence under section J.01.010;
(b) during the 10 years before the day on which the licence application is submitted, the applicant has contravened
(i) a provision of the Act, the Cannabis Act or their regulations, or
(ii) a term or condition of a licence or permit issued to the applicant under any regulations made under the Act or issued to the applicant under the Cannabis Act or its regulations;
(c) during the 10 years before the day on which the licence application is submitted the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subparagraph J.01.013(a)(i) or (b)(i) or received a sentence as specified in subparagraph J.01.013(a)(ii) or (b)(ii);
(d) an activity for which the licence is requested would contravene an international obligation;
(e) the applicant does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the licence is requested;
(f) the method referred to in paragraph J.01.014(1)(j) does not permit the recording of information as required under section J.01.075;
(g) the applicant has not complied with the requirements of subsection J.01.014(4) or the information and documents that they have provided are not sufficient to complete the review of the licence application;
(h) the Minister has reasonable grounds to believe that the applicant has submitted false or misleading information or false or falsified documents in or in support of the licence application;
(i) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the applicant has been involved in the diversion of a restricted drug to an illicit market or use or has been involved in an activity that contravened an international obligation; or
(j) the Minister has reasonable grounds to believe that the issuance of the licence would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to issue a licence under paragraph (1)(b) or (h) if the applicant meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the applicant does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the applicant has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to issue a licence, the Minister must send the applicant a notice that sets out the Minister’s reasons and gives the applicant an opportunity to be heard.
Renewal of Licence
Marginal note:Application
J.01.018 (1) To apply to renew a dealer’s licence, a licensed dealer must submit to the Minister an application that contains the information and documents referred to in subsections J.01.014(1) and (2).
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the senior person in charge of the site specified in the application; and
(b) include an attestation by that person that
(i) all of the information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the licensed dealer.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Renewal
J.01.019 (1) Subject to section J.01.021, on completion of the review of the renewal application, the Minister must issue a renewed dealer’s licence that contains the information specified in section J.01.015.
Marginal note:Terms and conditions
(2) When renewing a dealer’s licence, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to it or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence or the new level required as a result of the licence renewal; or
(c) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Validity
J.01.020 A renewed dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section J.01.035 or J.01.036.
Marginal note:Refusal
J.01.021 (1) The Minister must refuse to renew a dealer’s licence if
(a) the licensed dealer may no longer apply for a licence under section J.01.010;
(b) during the 10 years before the day on which the renewal application is submitted, the licensed dealer has contravened
(i) a provision of the Act, the Cannabis Act or their Regulations, or
(ii) a term or condition of a licence or permit issued to the dealer under a regulation made under the Act or issued to the dealer under the Cannabis Act or its regulations;
(c) during the 10 years before the day on which the renewal application is submitted, the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subparagraph J.01.013(a)(i) or (b)(i) or received a sentence as specified in subparagraph J.01.013(a)(ii) or (b)(ii);
(d) an activity for which the renewal is requested would contravene an international obligation;
(e) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the renewal is requested;
(f) the method referred to in paragraph J.01.014(1)(j) does not permit the recording of information as required under section J.01.075;
(g) the licensed dealer has not complied with the requirements of subsection J.01.018(3) or the information or documents that they have provided are not sufficient to complete the review of the renewal application;
(h) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the renewal application;
(i) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a restricted drug to an illicit market or use or has been involved in an activity that contravened an international obligation; or
(j) the Minister has reasonable grounds to believe that the renewal of the licence would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to renew a licence under paragraph (1)(b) or (h) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to renew a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
- SOR/85-550, s. 15
- SOR/2019-171, s. 22
Amendment of Licence
Marginal note:Application
J.01.022 (1) Before making a change affecting any information referred to in section J.01.015 that is contained in their dealer’s licence, a licensed dealer must submit to the Minister an application to amend the licence that contains a description of the proposed amendment, as well as the information and documents referred to in section J.01.014 that are relevant to the proposed amendment.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the senior person in charge of the site specified in the application; and
(b) include an attestation by that person that
(i) all of the information and documents submitted in support of the application are correct and complete to the best of their knowledge, and
(ii) they have the authority to bind the licensed dealer.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Amendment
J.01.023 (1) Subject to section J.01.025, on completion of the review of the amendment application, the Minister must amend the dealer’s licence.
Marginal note:Terms and conditions
(2) When amending a dealer’s licence, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to it or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence or the new level required as a result of the amendment; or
(c) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
- SOR/2004-238, s. 35
- SOR/2010-222, s. 31(F)
- SOR/2019-171, s. 22
Marginal note:Validity
J.01.024 An amended dealer’s licence is valid until the expiry date set out in the licence or, if it is earlier, the date of the suspension or revocation of the licence under section J.01.035 or J.01.036.
Marginal note:Refusal
J.01.025 (1) The Minister must refuse to amend a dealer’s licence if
(a) an activity for which the licence amendment is requested would contravene an international obligation;
(b) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of an activity for which the licence amendment is requested;
(c) the method referred to in paragraph J.01.014(1)(j) does not permit the recording of information as required by section J.01.075;
(d) the licensed dealer has not complied with the requirements of subsection J.01.022(3) or the information or documents that they have provided are not sufficient to complete the review of the amendment application;
(e) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the amendment application; or
(f) the Minister has reasonable grounds to believe that the amendment of the licence would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not refuse to amend a licence under paragraph (1)(e) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before refusing to amend a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
- SOR/2004-238, s. 36
- SOR/2010-222, s. 32
- SOR/2019-171, s. 22
Changes Requiring Prior Approval by Minister
Marginal note:Application
J.01.026 (1) A licensed dealer must obtain the Minister’s approval before making any of the following changes by submitting a written application to the Minister:
(a) a change affecting the security measures in place at the site specified in the dealer’s licence;
(b) the replacement of the senior person in charge;
(c) the replacement of the qualified person in charge; or
(d) the replacement or addition of an alternate qualified person in charge.
Marginal note:Information and documents
(2) The licensed dealer must provide the Minister with the following with respect to a change referred to in subsection (1):
(a) in the case of a change affecting the security measures in place at the site specified in the dealer’s licence, details of the change;
(b) in the case of the senior person in charge,
(i) the information specified in paragraph J.01.014(1)(c), and
(ii) the declaration specified in paragraph J.01.014(2)(b) and the documents specified in paragraphs J.01.014(2)(c) and (d); and
(c) in the case of the qualified person in charge or an alternate qualified person in charge,
(i) the information specified in paragraph J.01.014(1)(d), and
(ii) the declarations specified in paragraphs J.01.014(2)(b) and (e) and the documents specified in paragraphs J.01.014(2)(c), (d) and (f).
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
- SOR/2004-238, s. 37
- SOR/2019-171, s. 22
Marginal note:Approval
J.01.027 (1) Subject to section J.01.028, on completion of the review of the application for approval of the change, the Minister must approve the change.
Marginal note:Terms and conditions
(2) When approving a change, the Minister may, if he or she has reasonable grounds to believe that it is necessary to do so, add a term or condition to the licence or modify or delete one in order to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence; or
(c) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
- SOR/2004-238, s. 38
- SOR/2010-222, s. 33
- SOR/2019-171, s. 22
Marginal note:Refusal
J.01.028 (1) The Minister must refuse to approve the change if
(a) during the 10 years before the day on which the application for approval of the change is submitted, the proposed senior person in charge or qualified person in charge or any proposed alternate qualified person in charge was convicted as specified in subpa/ragraph J.01.013(a)(i) or (b)(i) or received a sentence as specified in subparagraph J.01.0013(a)(ii) or (b)(ii);
(b) the licensed dealer has not complied with the requirements of subsection J.01.026(3) or the information or documents that they have provided are not sufficient to complete the review of the application for approval of the change;
(c) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the application for approval of the change; or
(d) the Minister has reasonable grounds to believe that the change would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Exception
(2) The Minister must not refuse to approve a change under paragraph (1)(c) if the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use.
Marginal note:Notice
(3) Before refusing to approve a change, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard in respect of them.
- SOR/2015-210, s. 3
- SOR/2018-85, s. 2
- SOR/2019-171, s. 22
Changes Requiring Notice to Minister
Marginal note:Prior notice
J.01.029 (1) A licensed dealer must notify the Minister in writing before
(a) making or assembling a product or compound that is not set out in the most recent version of the list referred to in paragraph J.01.014(1)(f) that has been submitted to the Minister; or
(b) making a change to a product or compound that is set out in the list, if the change affects any of the information that has previously been submitted.
Marginal note:Information and list
(2) The notice must contain the information referred to in paragraph J.01.014(1)(f) that is necessary to update the list and be accompanied by the revised version of the list.
Marginal note:Notice — five days
J.01.030 A licensed dealer must notify the Minister in writing within five days after a person ceases to act as the qualified person in charge or an alternate qualified person in charge.
Marginal note:Notice — 10 days
J.01.031 (1) A licensed dealer must notify the Minister in writing within 10 days after one of the following changes occurs:
(a) a person ceases to act as the senior person in charge; or
(b) the licensed dealer ceases to manufacture or assemble a product or compound that is set out in the most recent version of the list referred to in paragraph J.01.014(1)(f) that has been submitted to the Minister.
Marginal note:Information and list
(2) A notice submitted under paragraph (1)(b) must specify which information referred to in paragraph J.01.014(1)(f) is being changed and be accompanied by the revised version of the list.
Marginal note:Notice of cessation of activities
J.01.032 (1) A licensed dealer that intends to cease conducting activities at their site — whether on or before the expiry of their licence — must notify the Minister in writing to that effect at least 30 days before ceasing those activities.
Marginal note:Content of notice
(2) The notice must be signed and dated by the senior person in charge and contain the following information:
(a) the expected date of the cessation of activities at the site;
(b) a description of the manner in which any remaining restricted drugs on the site as of that date will be disposed of by the licensed dealer, including
(i) if some or all of them will be sold or provided to another licensed dealer that will be conducting activities at the same site, the name of that dealer,
(ii) if some or all of them will be sold or provided to another licensed dealer that will not be conducting activities at the same site, the name of that dealer and the municipal address of their site, and
(iii) if some or all of them will be destroyed, the date on which and the municipal address of the location at which the destruction is to take place;
(c) the municipal address of the location at which the licensed dealer’s documents will be kept after activities have ceased; and
(d) the name, municipal address, telephone number and, if applicable, the email address of a person who the Minister may contact for further information after activities have ceased.
Marginal note:Update
(3) After having ceased to conduct the activities, the licensed dealer must submit to the Minister a detailed update of the information referred to in subsection (2) if it differs from what was set out in the notice. The update must be signed and dated by the senior person in charge.
- SOR/2004-238, s. 39
- SOR/2019-171, s. 22
J.01.032.1 [Repealed, SOR/2019-171, s. 22]
Changes to Terms and Conditions of Licence
Marginal note:Addition of or modification to term or condition
J.01.033 (1) The Minister may, at any time other than at the issuance, renewal or amendment of a dealer’s licence, add a term or condition to it or modify one if the Minister has reasonable grounds to believe that it is necessary to do so to
(a) ensure that an international obligation is respected;
(b) ensure conformity with the requirements associated with the security level specified in the licence; or
(c) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before adding a term or condition to a licence or modifying one, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Urgent circumstances
(3) Despite subsection (2), the Minister may add a term or condition to a licence or modify one without prior notice if the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use.
Marginal note:Urgent circumstances — notice
(4) The addition or modification of a term or condition that is made under subsection (3) takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the addition or modification;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
- SOR/99-125, s. 8
- SOR/2004-238, s. 40
- SOR/2018-69, ss. 67, 69
- SOR/2019-171, s. 22
J.01.033.1 [Repealed, SOR/2019-171, s. 22]
J.01.033.2 [Repealed, SOR/2019-171, s. 22]
J.01.033.3 [Repealed, SOR/2019-171, s. 22]
J.01.033.4 [Repealed, SOR/2019-171, s. 22]
Marginal note:Deletion of term or condition
J.01.034 (1) The Minister may delete a term or condition of a dealer’s licence that the Minister determines is no longer necessary.
Marginal note:Notice
(2) The deletion takes effect as soon as the Minister sends the licensed dealer a notice to that effect.
Suspension and Revocation of Licence
Marginal note:Suspension
J.01.035 (1) The Minister must suspend a dealer’s licence without prior notice if the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of licence
(3) The Minister must reinstate the licence if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
- SOR/97-228, s. 24
- SOR/2019-171, s. 22
Marginal note:Revocation
J.01.036 (1) Subject to subsection (2), the Minister must revoke a dealer’s licence if
(a) the licensed dealer may no longer apply for a licence under section J.01.010;
(b) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the licence or the actual or potential unauthorized use of the licence;
(c) the licensed dealer ceases to conduct activities at their site before the expiry of their licence;
(d) the licensed dealer does not take the corrective measures specified in an undertaking or notice;
(e) the licensed dealer has contravened
(i) a provision of the Act, the Cannabis Act or their regulations, or
(ii) a term or condition of a licence or permit issued to the dealer under a regulation made under the Act or issued to the dealer under the Cannabis Act or its regulations;
(f) during the 10 years before the day on which the licence is revoked, the senior person in charge, the qualified person in charge or any alternate qualified person in charge was convicted as specified in subparagraph J.01.013(a)(i) or (b)(i) or received a sentence as specified in subparagraph J.01.013(a)(ii) or (b)(ii);
(g) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of an application relating to the licence; or
(h) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a restricted drug to an illicit market or use.
Marginal note:Exceptions
(2) The Minister must not revoke a dealer’s licence for a ground set out in paragraph (1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking a licence, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
- SOR/97-228, s. 25
- SOR/2019-171, s. 22
Marginal note:Return of licence
J.01.037 The licensed dealer must return the original of the licence to the Minister within 15 days after the effective date of the revocation.
- SOR/2018-85, s. 3
- SOR/2019-171, s. 22
Import Permits
Marginal note:Application
J.01.038 (1) A licensed dealer must submit to the Minister, before each importation of a restricted drug, an application for an import permit that contains the following information:
(a) their name, municipal address and dealer’s licence number;
(b) with respect to the restricted drug to be imported,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt,
(iii) its quantity, and
(iv) in the case of a raw material, its purity and its anhydrous content;
(c) if the restricted drug is contained in a product to be imported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the restricted drug in the product, the number of units per package and the number of packages;
(d) the name and municipal address, in the country of export, of the exporter from whom the restricted drug is being obtained;
(e) the name of the customs office where the importation is anticipated; and
(f) each proposed mode of transportation and any proposed country of transit or transhipment.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that all of the information submitted in support of the application is correct and complete to the best of their knowledge.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
J.01.039 Subject to section J.01.042, on completion of the review of the import permit application, the Minister must issue to the licensed dealer an import permit that contains
(a) the permit number;
(b) the information set out in subsection J.01.038(1);
(c) the effective date of the permit;
(d) the expiry date of the permit, being the earlier of
(i) a date specified by the Minister that is not more than 180 days after its effective date, and
(ii) the expiry date of the dealer’s licence; and
(e) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected, or
(ii) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Validity
J.01.040 An import permit is valid until the earliest of
(a) the expiry date set out in the permit,
(b) the date of the suspension or revocation of the permit under section J.01.045 or J.01.046,
(c) the date of the suspension or revocation of the dealer’s licence under section J.01.035 or J.01.036, and
(d) the date of the suspension or revocation of the export permit that applies to the restricted drug to be imported and that is issued by the competent authority in the country of export.
Marginal note:Return of permit
J.01.041 If an import permit expires, the licensed dealer must return the original of the permit to the Minister within 15 days after its expiry.
Marginal note:Refusal
J.01.042 (1) The Minister must refuse to issue an import permit if
(a) the licensed dealer is not authorized by their dealer’s licence to import the relevant restricted drug or their licence will expire before the date of importation;
(b) the Minister has reasonable grounds to believe that the importation would contravene an international obligation;
(c) the licensed dealer does not have in place at the site the security measures set out in the Security Directive in respect of the importation;
(d) the licensed dealer has not complied with the requirements of subsection J.01.038(3) or the information or documents that they have provided are not sufficient to complete the review of the permit application;
(e) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the permit application;
(f) the licensed dealer has been notified that their application to renew or amend their licence will be refused;
(g) the Minister has reasonable grounds to believe that the importation would contravene the laws of the country of export or any country of transit or transhipment; or
(h) the Minister has reasonable grounds to believe that the issuance of the permit would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before refusing to issue the import permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Providing copy of permit
J.01.043 The holder of an import permit must provide a copy of the permit to the customs office at the time of importation.
Marginal note:Declaration
J.01.044 The holder of an import permit must provide the Minister, within 15 days after the day of release of the restricted drug specified in the permit in accordance with the Customs Act, with a declaration that contains the following information:
(a) their name and the numbers of their dealer’s licence and the import permit that applies to the restricted drug;
(b) with respect to the restricted drug,
(i) its name, as set out in the dealer’s licence,
(ii) if it is a salt, the name of the salt, and
(iii) its quantity;
(c) if the restricted drug is contained in a product that they have imported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the restricted drug in the product, the number of units per package and the number of packages; and
(d) the name of the customs office from which the restricted drug was released and the date of the release.
Marginal note:Suspension
J.01.045 (1) The Minister must suspend an import permit without prior notice if
(a) the dealer’s licence is suspended;
(b) the Minister has reasonable grounds to believe that the suspension is necessary to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use; or
(c) the importation would contravene the laws of the country of export or any country of transit or transhipment.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of permit
(3) The Minister must reinstate the import permit if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
Marginal note:Revocation
J.01.046 (1) Subject to subsection (2), the Minister must revoke an import permit if
(a) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the permit or the actual or potential unauthorized use of the permit;
(b) the licensed dealer does not carry out the corrective measures specified by the Minister under paragraph J.01.045(2)(c) by the specified date;
(c) the licensed dealer has contravened a term or condition of the permit;
(d) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of the application for the permit;
(e) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a restricted drug to an illicit market or use; or
(f) the dealer’s licence has been revoked.
Marginal note:Exceptions
(2) The Minister must not revoke an import permit for a ground set out in paragraph (1)(d) or J.01.036(1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking an import permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Return of permit
J.01.047 If an import permit is revoked, the licensed dealer must return the original of the permit to the Minister within 15 days after the effective date of the revocation.
Export Permits
Marginal note:Application
J.01.048 (1) A licensed dealer must submit to the Minister, before each exportation of a restricted drug, an application for an export permit that contains the following information and document:
(a) their name, municipal address and dealer’s licence number;
(b) with respect to the restricted drug to be exported,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt,
(iii) its quantity, and
(iv) in the case of a raw material, its purity and its anhydrous content;
(c) if the restricted drug is contained in a product to be exported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the restricted drug in the product, the number of units per package and the number of packages;
(d) the name and municipal address of the importer in the country of final destination;
(e) the name of the customs office where the exportation is anticipated;
(f) each proposed mode of transportation and any proposed country of transit or transhipment; and
(g) a copy of the import permit issued by the competent authority in the country of final destination that sets out the name of the importer and the municipal address of their site in that country.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that, to the best of their knowledge,
(i) the exportation does not contravene the laws of the country of final destination or any country of transit or transhipment, and
(ii) all of the information and documents submitted in support of the application are correct and complete.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Issuance
J.01.049 Subject to section J.01.052, on completion of the review of the export permit application, the Minister must issue to the licensed dealer an export permit that contains
(a) the permit number;
(b) the information set out in paragraphs J.01.048(1)(a) to (f);
(c) the effective date of the permit;
(d) the expiry date of the permit, being the earliest of
(i) a date specified by the Minister that is not more than 180 days after its effective date,
(ii) the expiry date of the dealer’s licence, and
(iii) the expiry date of the import permit issued by the competent authority in the country of final destination; and
(e) any terms and conditions that the Minister has reasonable grounds to believe are necessary to
(i) ensure that an international obligation is respected, or
(ii) reduce a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Validity
J.01.050 An export permit is valid until the earliest of
(a) the expiry date set out in the permit,
(b) the date of the suspension or revocation of the permit under section J.01.055 or J.01.056,
(c) the date of the suspension or revocation of the dealer’s licence under section J.01.035 or J.01.036, and
(d) the date of the expiry, suspension or revocation of the import permit that applies to the restricted drug to be exported and that is issued by the competent authority in the country of final destination.
Marginal note:Return of permit
J.01.051 If an export permit expires, the licensed dealer must return the original of the permit to the Minister within 15 days after its expiry.
Marginal note:Refusal
J.01.052 (1) The Minister must refuse to issue an export permit if
(a) the licensed dealer is not authorized by their dealer’s licence to export the relevant restricted drug or their dealer’s licence will expire before the date of export;
(b) the Minister has reasonable grounds to believe that the exportation would contravene an international obligation;
(c) the licensed dealer has not complied with the requirements of subsection J.01.048(3) or the information or documents that they have provided are not sufficient to complete the review of the permit application;
(d) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the permit application;
(e) the licensed dealer has been notified that their application to renew or amend their licence will be refused;
(f) the Minister has reasonable grounds to believe that the exportation would not be in conformity with the import permit issued by the competent authority of the country of final destination;
(g) the Minister has reasonable grounds to believe that the exportation would contravene the laws of the country of final destination or any country of transit or transhipment; or
(h) the Minister has reasonable grounds to believe that the issuance of the permit would likely create a risk to public health or safety, including the risk of a restricted drug being diverted to an illicit market or use.
Marginal note:Notice
(2) Before refusing to issue the export permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Providing copy of permit
J.01.053 The holder of an export permit must provide a copy of the permit to the customs office at the time of exportation.
Marginal note:Declaration
J.01.054 The holder of an export permit must provide the Minister, within 15 days after the day of export of the restricted drug specified in the permit, with a declaration that contains the following information:
(a) their name and the numbers of their dealer’s licence and the export permit that applies to the restricted drug;
(b) with respect to the restricted drug,
(i) its name, as specified in the dealer’s licence,
(ii) if it is a salt, the name of the salt, and
(iii) its quantity;
(c) if the restricted drug is contained in a product that they have exported,
(i) the brand name of the product,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any, and
(iii) the strength per unit of the restricted drug in the product, the number of units per package and the number of packages; and
(d) the name of the customs office from which the restricted drug was exported and the date of export.
Marginal note:Suspension
J.01.055 (1) The Minister must suspend an export permit without prior notice if
(a) the dealer’s licence is suspended;
(b) the Minister has reasonable grounds to believe that the suspension is necessary to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use; or
(c) the exportation would contravene the laws of the country of final destination or any country of transit or transhipment.
Marginal note:Notice
(2) The suspension takes effect as soon as the Minister sends the licensed dealer a notice that
(a) sets out the reasons for the suspension;
(b) gives the dealer an opportunity to be heard; and
(c) if applicable, specifies the corrective measures that must be carried out and the date by which they must be carried out.
Marginal note:Reinstatement of permit
(3) The Minister must reinstate the export permit if the Minister has reasonable grounds to believe that the suspension is no longer necessary.
Marginal note:Revocation
J.01.056 (1) Subject to subsection (2), the Minister must revoke an export permit if
(a) the licensed dealer requests the Minister to do so or informs the Minister of the loss or theft of the permit or the actual or potential unauthorized use of the permit;
(b) the licensed dealer does not carry out the corrective measures specified by the Minister under paragraph J.01.055(2)(c) by the specified date;
(c) the licensed dealer has contravened a term or condition of the permit;
(d) the Minister has reasonable grounds to believe that the licensed dealer submitted false or misleading information or false or falsified documents in or in support of the application for the permit;
(e) information received from a competent authority or the United Nations gives the Minister reasonable grounds to believe that the licensed dealer has been involved in the diversion of a restricted drug to an illicit market or use; or
(f) the dealer’s licence has been revoked.
Marginal note:Exceptions
(2) The Minister must not revoke an export permit for a ground set out in paragraph (1)(d) or J.01.036(1)(e) or (g) if the licensed dealer meets the following conditions, unless the Minister has reasonable grounds to believe that it is necessary to do so to protect public health or safety, including to prevent a restricted drug from being diverted to an illicit market or use:
(a) the licensed dealer does not have a history of non-compliance with the Act, the Cannabis Act or their regulations; and
(b) the licensed dealer has carried out, or signed an undertaking to carry out, the necessary corrective measures to ensure compliance with the Act, the Cannabis Act and their regulations.
Marginal note:Notice
(3) Before revoking an export permit, the Minister must send the licensed dealer a notice that sets out the Minister’s reasons and gives the dealer an opportunity to be heard.
Marginal note:Return of permit
J.01.057 If an export permit is revoked, the licensed dealer must return the original of the permit to the Minister within 15 days after the effective date of the revocation.
Identification
Marginal note:Name
J.01.058 A licensed dealer must include their name, as set out in their dealer’s licence, on all the means by which they identify themself in regard to their activities in relation to restricted drugs, including labels, orders, shipping documents, invoices and advertising.
Sale of Restricted Drugs
Marginal note:Sale to institution
J.01.059 (1) Despite section C.08.002 and subject to subsections (3) and (4), a licensed dealer may sell a restricted drug to an institution for one of the following purposes if the institution submits to the dealer or the Minister an application to purchase the drug and the Minister issues a prior written authorization for the sale:
(a) clinical testing in the institution by qualified investigators for the purpose of determining the hazards and efficacy of the drug; or
(b) laboratory research in the institution by qualified investigators.
Marginal note:Content of application
(2) The application must contain the following information:
(a) the name and the municipal address of the institution;
(b) the names and qualifications of the qualified investigators;
(c) the name, form, quantity and strength per unit of the restricted drug being requested;
(d) details of the proposed use of the drug; and
(e) the name and municipal address of the licensed dealer from whom the institution proposes to purchase the drug.
Marginal note:Application to licensed dealer
(3) If the institution submits the application to the licensed dealer, the dealer must provide a copy of it to the Minister.
Marginal note:Authorization by Minister
(4) After reviewing the application received from the institution or the copy of it received from the licensed dealer, the Minister may, subject to any terms and conditions that the Minister has reasonable grounds to believe are necessary, authorize in writing
(a) the sale by the licensed dealer to the institution of the restricted drug applied for in the form, quantity and strength per unit specified by the Minister; and
(b) the possession of the restricted drug by qualified investigators for clinical testing of the drug in the institution for the purpose of determining its hazards and efficacy or to conduct laboratory research with the drug in the institution.
Marginal note:Authorized use only
(5) The institution must use the restricted drug only in accordance with the written authorization.
Marginal note:Sale to Minister
J.01.060 A licensed dealer may sell or provide a restricted drug to the Minister.
Marginal note:Provision for identification or analysis
J.01.061 (1) Despite anything in this Part, a person may, for the purpose of identification or analysis of a restricted drug, provide or deliver it to
(a) a practitioner of medicine; or
(b) an agent or mandatary of a practitioner of medicine, if the agent or mandatary has been exempted under section 56 of the Act with respect to the possession of that restricted drug for that purpose.
Marginal note:Agent or mandatary of practitioner of medicine
(2) An agent or mandatary of a practitioner of medicine who receives the restricted drug must immediately provide or deliver it to
(a) the practitioner; or
(b) the Minister.
Marginal note:Practitioner of medicine
(3) A practitioner of medicine who receives the restricted drug must immediately provide or deliver it
(a) for the purpose of its identification or analysis, to a person exempted under section 56 of the Act with respect to the possession of that restricted drug for that purpose; or
(b) to the Minister.
Packaging, Labelling and Transportation
Marginal note:Packaging — sale and provision
J.01.062 (1) A licensed dealer that sells or provides a restricted drug must securely package it in its immediate container, which must be sealed in such a manner that the container cannot be opened without breaking the seal.
Marginal note:Packaging — transport and export
(2) A licensed dealer that transports or exports a restricted drug must ensure that its package is sealed in such a manner that the package cannot be opened without breaking the seal.
Marginal note:Exception
(3) Subsection (1) does not apply to a test kit that contains a restricted drug and that has a registration number.
Marginal note:Labelling
J.01.063 (1) A licensed dealer that sells or provides a restricted drug must ensure that its package is labelled so that its inner and outer labels show
(a) the proper name or, if there is no proper name, the name of the drug;
(b) the net contents of the package;
(c) the unit strength of the drug and the number of units per package, if applicable;
(d) the lot number of the drug;
(e) the expression “Restricted Drug”; and
(f) the name and municipal address of the manufacturer or assembler of the drug.
Marginal note:Exception
(2) Subsection (1) does not apply to a test kit that contains a restricted drug and that has a registration number.
Marginal note:Non-application
(3) The labelling requirements set out in section C.01.004 do not apply to a restricted drug.
Marginal note:Transport
J.01.064 A licensed dealer must, in taking delivery of a restricted drug that they have imported or in making delivery of a restricted drug,
(a) take any measures that are necessary to ensure the security of the drug while it is being transported;
(b) use a method of transportation that permits an accurate record to be kept of all handling of the drug as well as of the signatures of every person handling it until it is delivered to the consignee;
(c) in the case of an imported drug, transport it directly to the site specified in their licence after it is released under the Customs Act; and
(d) in the case of a drug to be exported, transport it directly from the site specified in their licence to the customs office where it will be exported.
Thefts, Losses and Suspicious Transactions
Marginal note:Protective measures — licences and permits
J.01.065 A licensed dealer must take any measures that are necessary to ensure the security of any licence or permit in their possession.
Marginal note:Protective measures — restricted drugs
J.01.066 The following persons must take any measures that are necessary to ensure the security of any restricted drugs in their possession:
(a) a licensed dealer;
(b) an institution;
(c) a qualified investigator who possesses the restricted drug for the purpose of clinical testing or laboratory research in an institution; and
(d) a person exempted under section 56 of the Act with respect to the possession of the restricted drug.
Marginal note:Theft or loss — licences and permits
J.01.067 A licensed dealer that becomes aware of a theft or loss of their licence or permit must provide a written report to the Minister within 72 hours after becoming aware of it.
Marginal note:Theft or loss — restricted drugs
J.01.068 (1) Subject to subsection (2), any person referred to in section J.01.066 who becomes aware of a theft or loss of a restricted drug must
(a) provide a written report to a member of a police force within 24 hours after becoming aware of the theft or loss; and
(b) provide a written report to the Minister within 72 hours after becoming aware of the theft or loss and include a confirmation that the report required under paragraph (a) has been provided.
Marginal note:Explainable loss — licensed dealer
(2) Subsection (1) does not apply to a licensed dealer that becomes aware of a loss of a restricted drug that can be explained on the basis of normally accepted business activities.
Marginal note:Suspicious transaction
J.01.069 (1) A licensed dealer must provide a written report containing the following information to the Minister within 72 hours after becoming aware of a transaction occurring in the course of their activities that they have reasonable grounds to suspect may be related to the diversion of a restricted drug to an illicit market or use:
(a) their name, municipal address, telephone number and, if the licensed dealer is a corporation, the position held by the individual making the report;
(b) the name and municipal address of the other party to the transaction;
(c) details of the transaction, including its date and time, its type, the name and quantity of the restricted drug and, in the case of a product or compound, the quantity of every restricted drug that it contains;
(d) in the case of a product that contains the restricted drug, other than a test kit, the drug identification number that is assigned to the product under section C.01.014.2, if any; and
(e) a detailed description of the reasons for those suspicions.
Marginal note:Good faith
(2) No civil proceedings lie against a licensed dealer for having provided the report in good faith.
Marginal note:Non-disclosure
(3) A licensed dealer must not disclose that they have provided the report or disclose details of it, with the intent to prejudice a criminal investigation, whether or not a criminal investigation has begun.
Marginal note:Partial protection against self-incrimination
J.01.070 A report made under any of sections J.01.067 to J.01.069, or any evidence derived from it, is not to be used or received to incriminate the licensed dealer in any criminal proceeding against them other than a prosecution under section 132, 136 or 137 of the Criminal Code.
Destruction of Restricted Drugs
Marginal note:Destruction at site
J.01.071 A licensed dealer that intends to destroy a restricted drug at the site specified in their licence must ensure that the following conditions are met:
(a) the licensed dealer obtains the prior approval of the Minister;
(b) the destruction occurs in the presence of two of the following persons, at least one of whom must be a person referred to in subparagraph (i):
(i) the senior person in charge, the qualified person in charge or an alternate qualified person in charge, and
(ii) a person who works for or provides services to the licensed dealer and holds a senior position;
(c) the destruction is carried out in accordance with a method that complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction; and
(d) as soon as the destruction is completed, the person who carried out the destruction and each of the two persons referred to in paragraph (b) who were present at the destruction sign and date a joint declaration attesting that the restricted drug was completely destroyed, to which each signatory must add their name in printed letters.
Marginal note:Destruction elsewhere than at site
J.01.072 A licensed dealer that intends to destroy a restricted drug elsewhere than at the site specified in their licence must ensure that the following conditions are met:
(a) the licensed dealer obtains the prior approval of the Minister;
(b) the licensed dealer takes any measures that are necessary to ensure the security of the restricted drug while it is being transported in order to prevent its diversion to an illicit market or use;
(c) the destruction is carried out by a person working for a business that specializes in the destruction of dangerous goods and in the presence of another person working for that business;
(d) the destruction is carried out in accordance with a method that complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction; and
(e) as soon as the destruction is completed, the person who carried out the destruction provides the licensed dealer with a dated declaration attesting that the restricted drug was completely destroyed and containing
(i) the municipal address of the place of destruction,
(ii) the name and quantity of the restricted drug and, if applicable, the brand name and quantity of the product containing it or the name and quantity of the compound containing it,
(iii) the method of destruction,
(iv) the date of destruction, and
(v) the names in printed letters and signatures of that person and the other person who was present at the destruction.
Marginal note:Application for prior approval
J.01.073 (1) A licensed dealer must submit to the Minister an application that contains the following information in order to obtain the Minister’s prior approval to destroy a restricted drug:
(a) their name, municipal address and dealer’s licence number;
(b) the proposed date of destruction;
(c) the municipal address of the place of destruction;
(d) a brief description of the method of destruction;
(e) if the destruction is to be carried out at the site specified in the dealer’s licence, the names of the persons proposed for the purpose of paragraph J.01.071(b) and information establishing that they meet the conditions of that paragraph;
(f) the name of the restricted drug and, if applicable, the brand name of the product containing it or the name of the compound containing it; and
(g) the form and quantity of the restricted drug or the product or compound containing it and if applicable, the strength per unit of the restricted drug in the product or compound, the number of units per package and the number of packages.
Marginal note:Signature and attestation
(2) The application must
(a) be signed and dated by the qualified person in charge or an alternate qualified person in charge; and
(b) include an attestation by that person that
(i) the proposed method of destruction complies with all federal, provincial and municipal environmental protection legislation applicable to the place of destruction, and
(ii) all of the information submitted in support of the application is correct and complete to the best of the signatory’s knowledge.
Marginal note:Additional information and documents
(3) The licensed dealer must, not later than the date specified in the Minister’s written request to that effect, provide the Minister with any information or document that the Minister determines is necessary to complete the review of the application.
Marginal note:Approval
J.01.074 On completion of the review of the application for approval, the Minister must approve the destruction of the restricted drug unless
(a) in the case of a destruction that is to be carried out at the site specified in the dealer’s licence, the persons proposed for the purpose of paragraph J.01.071(b) do not meet the conditions of that paragraph;
(b) the Minister has reasonable grounds to believe that the restricted drug would not be destroyed;
(c) the Minister has reasonable grounds to believe that the licensed dealer has submitted false or misleading information or false or falsified documents in or in support of the approval application;
(d) the restricted drug or a portion of it is required for the purposes of a criminal or administrative investigation or a preliminary inquiry, trial or other proceeding under any Act or its regulations; or
(e) the Minister has reasonable grounds to believe that the approval would likely create a risk to public health or safety, including the risk of the restricted drug being diverted to an illicit market or use.
Documents
Licensed Dealers
Marginal note:Method of recording information
J.01.075 A licensed dealer must record any information that they are required to record under this Part using a method that permits an audit of it to be made at any time.
Marginal note:Information — general
J.01.076 A licensed dealer must record the following information:
(a) the name, form and quantity of any restricted drug that the dealer orders, the name of the person who placed the order on the dealer’s behalf and the date of the order;
(b) the name, form and quantity of any restricted drug that the dealer receives, the name and municipal address of the person who sold or provided it and the date on which it was received;
(c) in the case of a restricted drug that the dealer sells or provides,
(i) the brand name of the product or the name of the compound containing the restricted drug and the name of the restricted drug,
(ii) the drug identification number that has been assigned to the product under section C.01.014.2, if any,
(iii) the form and quantity of the restricted drug and, if applicable, the strength per unit of the restricted drug in the product or compound, the number of units per package and the number of packages,
(iv) the name and municipal address of the person to whom it was sold or provided, and
(v) the date on which it was sold or provided;
(d) the name, form and quantity of any restricted drug that the dealer manufactures or assembles and the date on which it was placed in stock and, if applicable, the strength per unit of the restricted drug in the product or compound, the number of units per package and the number of packages;
(e) the name and quantity of any restricted drug that the dealer uses in the manufacturing or assembling of a product or compound, as well as the brand name and quantity of the product or the name and quantity of the compound, and the date on which the product or compound was placed in stock;
(f) the name, form and quantity of any restricted drug in stock at the end of each month;
(g) the name, form and quantity of any restricted drug that the dealer delivers, transports or sends, the name and municipal address of the consignee and the date on which it was delivered, transported or sent;
(h) the name, form and quantity of any restricted drug imported, the date on which it was that the dealer imports, the name and municipal address of the exporter, the country of exportation and any country of transit or transhipment; and
(i) the name, form and quantity of any restricted drug that the dealer exports, the date on which it was exported, the name and municipal address of the importer, the country of final destination and any country of transit or transhipment.
Marginal note:Explainable loss of restricted drug
J.01.077 A licensed dealer that becomes aware of a loss of a restricted drug that can be explained on the basis of normally accepted business activities must record the following information:
(a) the name of the lost restricted drug and, if applicable, the brand name of the product or the name of the compound containing it;
(b) the form and quantity of the restricted drug and, if applicable, the form of the product or compound containing it, the strength per unit of the restricted drug in the product or compound, the number of units per package and the number of packages;
(c) the date on which the dealer became aware of the loss; and
(d) the explanation for the loss.
Marginal note:Destruction
J.01.078 A licensed dealer must record the following information concerning any restricted drug that they destroy at the site specified in their licence:
(a) the municipal address of the place of destruction;
(b) the name, form and quantity of the restricted drug and, if applicable, the brand name and quantity of the product containing the drug or the name and quantity of the compound containing the drug;
(c) the method of destruction; and
(d) the date of destruction.
Marginal note:Annual report
J.01.079 (1) Subject to subsections (2) and (3), a licensed dealer must provide to the Minister, within three months after the end of each calendar year, an annual report that contains
(a) the name, form and total quantity of each restricted drug that they receive, produce, sell, provide, import, export or destroy during the calendar year, as well as the name and total quantity of each restricted drug that they use to manufacture or assemble a product or compound;
(b) the name, form and quantity of each restricted drug in physical inventory taken at the site specified in their licence at the end of the calendar year; and
(c) the name, form and quantity of any restricted drug that has been lost in the course of conducting activities during the calendar year.
Marginal note:Non-renewal or revocation within first three months
(2) If a licensed dealer’s licence expires without being renewed or is revoked during the first three months of a calendar year, the dealer must provide to the Minister
(a) within three months after the end of the preceding calendar year, the annual report in respect of that year; and
(b) within three months after the expiry or revocation, a report in respect of the portion of the current calendar year during which the licence was valid that contains the information referred to in subsection (1), in which the quantity in physical inventory is to be calculated as of the date of expiry or revocation.
Marginal note:Non-renewal or revocation after third month
(3) If a licensed dealer’s licence expires without being renewed or is revoked after the first three months of a calendar year, the dealer must provide to the Minister, within three months after the expiry or revocation, a report in respect of the portion of the calendar year during which the licence was valid that contains the information referred to in subsection (1) for that period, in which the quantity in physical inventory is to be calculated as of the date of expiry or revocation.
Institutions
Marginal note:Method of recording information
J.01.080 An institution must record any information that it is required to record under this Part using a method that permits an audit of the information to be made at any time.
Marginal note:Information
J.01.081 An institution must record the following information:
(a) the name and quantity of any restricted drug that the institution orders, the name of the person who placed the order on the institution’s behalf and the date of the order;
(b) the name and quantity of any restricted drug that the institution receives as well as the name and municipal address of the licensed dealer that sold or provided it and the date on which it was received;
(c) details of the use of restricted drugs in the institution;
(d) the names and qualifications of every person who makes use of a restricted drug in the institution; and
(e) all clinical data with respect to the use of every restricted drug received by the institution.
Drug Received for Identification and Analysis
Marginal note:Method of recording information
J.01.082 A person who records information in accordance with section J.01.083 must do so using a method that permits an audit of the information to be made at any time.
Marginal note:Information
J.01.083 A person who receives a restricted drug in accordance with section J.01.061 must record the following information:
(a) the name and quantity of the restricted drug, as well as the name and municipal address of the person who provided it to them and the date on which it was received;
(b) details regarding the identification or analysis of the restricted drug; and
(c) the names of the persons who handled the restricted drug during the process of identifying or analyzing it.
Record Keeping
Marginal note:Retention period
J.01.084 A licensed dealer, a former licensed dealer, an institution and a person referred to in section J.01.083 must keep any document containing the information that they are required to record under this Part, including every declaration and a copy of every report, for a period of two years following the day on which the last record is recorded in the document and in a manner that permits an audit of the document to be made at any time.
Marginal note:Location
J.01.085 The documents must be kept
(a) in the case of a licensed dealer, at the site specified in their licence;
(b) in the case of an institution, at the institution;
(c) in the case of a person referred to in section J.01.083, at a location in Canada; and
(d) in the case of a former licensed dealer, at a location in Canada.
Marginal note:Quality of documents
J.01.086 The documents must be complete and readily retrievable and the information in them must be legible and indelible.
Notification of Application for Order of Restoration
Marginal note:Written notification
J.01.087 (1) For the purpose of subsection 24(1) of the Act, notification of an application for an order of restoration must be given in writing to the Attorney General by registered mail and be mailed at least 15 days before the date on which the application is to be made to a justice.
Marginal note:Content of notification
(2) The notification must specify
(a) the name of the justice to whom the application is to be made;
(b) the time and place at which the application is to be heard;
(c) details concerning the restricted drug or other thing in respect of which the application is to be made; and
(d) the evidence on which the applicant intends to rely to establish that they are entitled to possession of the restricted drug or other thing referred to in paragraph (c).
SCHEDULE(Sections J.01.001, J.01.002 and J.01.004)
PART I
- 1The following amphetamines, their salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues:
- (1)N-ethylamphetamine (N-ethyl-α-methylbenzeneethanamine)
- (2)4-methyl-2,5-dimethoxyamphetamine (STP) (2,5-dimethoxy-4,α-dimethylbenzeneethanamine)
- (3)3,4-methylenedioxyamphetamine (MDA) (α-methyl-1,3-benzodioxole-5-ethanamine)
- (4)2,5-dimethoxyamphetamine(2,5-dimethoxy-α-methylbenzeneethanamine)
- (5)4-methoxyamphetamine (4-methoxy-α-methylbenzeneethanamine)
- (6)2,4,5-trimethoxyamphetamine (2,4,5-trimethoxy-α-methylbenzeneethanamine)
- (7)N-methyl-3,4-methylenedioxyamphetamine (N,α-dimethyl-1,3-benzodioxole-5-ethanamine)
- (8)4-ethoxy-2,5-dimethoxyamphetamine (4-ethoxy-2,5-dimethoxy-α-methylbenzeneethanamine)
- (9)5-methoxy-3,4-methylenedioxyamphetamine (7-methoxy-α-methyl-1,3-benzodioxole-5-ethanamine)
- (10)N,N-dimethyl-3,4-methylenedioxyamphetamine (N,N, α-trimethyl-1,3-benzodioxole-5-ethanamine)
- (11)N-ethyl-3,4-methylenedioxyamphetamine (N-ethyl-α-methyl-1,3-benzodioxole-5-ethanamine)
- (12)4-ethyl-2,5-dimethoxyamphetamine (DOET) (4-ethyl-2,5-dimethoxy-α-methylbenzeneethanamine)
- (13)4-bromo-2,5-dimethoxyamphetamine (4-bromo-2,5-dimethoxy-α-methylbenzeneethanamine)
- (14)4-chloro-2,5-dimethoxyamphetamine (4-chloro-2,5-dimethoxy-α-methylbenzeneethanamine)
- (15)4-ethoxyamphetamine (4-ethoxy-α-methyl-benzeneethanamine)
- (16)N-Propyl-3,4-methylenedioxyamphetamine (α-methyl-N-propyl-1,3-benzodioxole-5-ethanamine)
- (17)N-hydroxy-3,4-methylenedioxyamphetamine (N-[α-methyl-3,4-(methylenedioxy)phenethyl]hydroxylamine)
- (18)3,4,5-trimethoxyamphetamine (3,4,5-trimethoxy-α-methylbenzeneethanamine)
- 2Lysergic acid diethylamide (LSD) (N,N-diethyllysergamide) and any salt thereof
- 3N,N-Diethyltryptamine (DET) (3-[(2-diethylamino)ethyl]indole) and any salt thereof
- 4N,N-Dimethyltryptamine (DMT) (3-[(2-dimethylamino)ethyl]indole) and any salt thereof
- 5N-Methyl-3-piperidyl benzilate (LBJ) (3-[(hydroxydiphenylacetyl)oxy]-1-methylpiperidine) and any salt thereof
- 6Harmaline (4,9-dihydro-7-methoxy-1-methyl-3H-pyrido(3,4-b)indole) and any salt thereof
- 7Harmalol (4,9-dihydro-1-methyl-3H-pyrido(3,4-ß)indol-7-ol) and any salt thereof
- 8Psilocin (3-[2-(dimethylamino)ethyl]-4-hydroxyindole) and any salt thereof
- 9Psilocybin (3-[2-(dimethylamino)ethyl]-4-phosphoryloxyindole) and any salt thereof
- 10N-(1-phenylcyclohexyl)ethylamine (PCE) and any salt thereof
- 111-[1-(2-Thienyl)cyclohexyl]piperidine (TCP) and any salt thereof
- 121-Phenyl-N-propylcyclohexanamine and any salt thereof
- 13Mescaline (3,4,5-trimethoxybenzeneethanamine) and any salt thereof, but not peyote (lophophora)
- 14[Repealed, SOR/2017-250, s. 2]
- 152-Methylamino-1-phenyl-1-propanone and any salt thereof
- 161-[1-(Phenylmethyl)cyclohexyl]piperidine and any salt thereof
- 171-[1-(4-Methylphenyl)cyclohexyl]piperidine and any salt thereof
- 18Etryptamine (3-(2-aminobutyl)indole) and any salt thereof
- 19Rolicyclidine (1-(1-phenylcyclohexyl) pyrrolidine) and any salt thereof
- 20Benzylpiperazine [BZP], namely 1-benzylpiperazine and its salts, isomers and salts of isomers
- 21Trifluoromethylphenylpiperazine [TFMPP], namely 1-(3-trifluoromethylphenyl)piperazine and its salts, isomers and salts of isomers
- 22Methylenedioxypyrovalerone (MDPV), its salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues
- 23Cathinone ((-)-α-aminopropiophenone) and its salts
- 242C-phenethylamines and their salts, derivatives, isomers and salts of derivatives and isomers that correspond to the following chemical description:
- any substance that has a 1-amino-2-phenylethane structure substituted at the 2’ and 5’ or 2’ and 6’ positions of the benzene ring by an alkoxy or haloalkoxy group, or substituted at two adjacent carbon atoms of the benzene ring which results in the formation of a furan, dihydrofuran, pyran, dihydropyran or methylenedioxy group — whether or not further substituted on the benzene ring to any extent, whether or not substituted at the amino group by one or two, or a combination of, methyl, ethyl, propyl, isopropyl, hydroxyl, benzyl (or benzyl substituted to any extent) or benzylene (or benzylene substituted to any extent) groups and whether or not substituted at the 2-ethyl (beta carbon) position by a hydroxyl, oxo or alkoxy group — and its salts and derivatives and salts of derivatives, including
- (1)4-bromo-2,5-dimethoxy-N-(2-methoxybenzyl)phenethylamine (25B-NBOMe)
- (2)4-chloro-2,5-dimethoxy-N-(2-methoxybenzyl)phenethylamine (25C-NBOMe)
- (3)4-iodo-2,5-dimethoxy-N-(2-methoxybenzyl)phenethylamine (25I-NBOMe)
- (4)4-bromo-2,5-dimethoxybenzeneethanamine (2C-B)
- 25AH-7921 (1-(3,4-dichlorobenzamidomethyl)cyclohexyldimethylamine), its salts, isomers and salts of isomers
- 26MT-45 (1-cyclohexyl-4-(1,2-diphenylethyl)piperazine), its salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues, including
- (1)Diphenidine (DEP) (1-(1,2-diphenylethyl)piperidine)
- (2)Methoxphenidine (2-MeO-Diphenidine, MXP) (1-[1-(2-methoxyphenyl)-2-phenylethyl]piperidine)
- (3)Ephenidine (NEDPA, EPE) (N-ethyl-1,2-diphenylethylamine)
- (4)Isophenidine (NPDPA) (N-isopropyl-1,2-diphenylethylamine)
- but not including
- (5)Lefetamine ((-)-N,N-dimethyl-α-phenylbenzeneethanamine), its salts, derivatives and isomers and salts of derivatives and isomers
- 27W-18 (4-chloro-N-[1-[2-(4-nitrophenyl)ethyl]-2-piperidinylidene]benzenesulfonamide), its salts, derivatives, isomers and analogues and salts of derivatives, isomers and analogues
- 28U-47700 (3,4-dichloro-N-(2-(dimethylamino)cyclohexyl)-N-methylbenzamide), its salts, derivatives, isomers and analogues, and salts of derivatives, isomers and analogues, including
- (1)Bromadoline (4-bromo-N-(2-(dimethylamino)cyclohexyl)benzamide)
- (2)U-47109 (3,4-dichloro-N-(2-(dimethylamino)cyclohexyl)benzamide)
- (3)U-48520 (4-chloro-N-(2-(dimethylamino)cyclohexyl)-N-methylbenzamide)
- (4)U-50211 (N-(2-(dimethylamino)cyclohexyl)-4-hydroxy-N-methylbenzamide)
- (5)U-77891 (3,4-dibromo-N-methyl-N-(1-methyl-1-azaspiro[4.5]decan-6-yl)benzamide)
PART II
- 1Salvia divinorum (S. divinorum), its preparations and derivatives, including
- (1)Salvinorin A ((2S,4aR,6aR,7R,9S,10aS,10bR)-9-(acetyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho[2,1-c]pyran-7-carboxylic acid methyl ester)
- 2Catha edulis Forsk, its preparations, derivatives, alkaloids and salts, including
- (1)Cathine (d-threo-2-amino-1-hydroxy-1-phenylpropane)
- but not including
- (2)Cathinone ((-)-α-aminopropiophenone) and its salts
| Item | Column 1 | Column 2 |
|---|---|---|
| Substance | Period | |
- SOR/97-228, s. 25
- SOR/2003-34, ss. 4, 5
- SOR/2012-65, s. 1
- SOR/2012-177, s. 1
- SOR/2013-172, s. 2
- SOR/2015-210, ss. 4 to 6
- SOR/2016-72, s. 1
- SOR/2016-106, s. 2
- SOR/2016-239, s. 1
- SOR/2017-12, ss. 3, 4
- SOR/2017-250, s. 2
- SOR/2017-278, s. 1
- SOR/2018-69, s. 66
- SOR/2018-85, s. 4
- SOR/2019-171, s. 23
SCHEDULE F
SCHEDULE KReasonable Daily Intake for Various Foods
| Column I | Column II | ||
|---|---|---|---|
| Item No. | Name and Description | R.D.I. | |
| 1 | Alimentary Pastes, dry | 3.0 oz. | 85 g |
| 2 | Bacon (side) simulated meat product that resembles side bacon, (cooked) | 1.0 oz. | 28 g |
| 3 | Beverage Bases and Mixes, Flavoured, for Addition to Milk (ready to serve) | 16.0 fl.oz. | 454 ml |
| 4 | Bread, 5 slices | 5.3 oz. | 150 g |
| 5 | Butter | 2.0 oz. | 57 g |
| 6 | Buttermilk | 30.0 fl.oz. | 852 ml |
| 7 | Cereals, Breakfast or Infant | 1.0 oz. | 28 g |
| 8 | Cereals, puffed | 0.5 oz. | 14 g |
| 9 | Cheese (other than Cottage Cheese) | 2.0 oz. | 57 g |
| 10 | Cheese, Cottage | 3.5 oz. | 100 g |
| 11 | Condensed Milk | 15.0 fl.oz. | 426 ml |
| 12 | Cream, whipping | 2.0 oz. | 57 g |
| 13 | Egg, yolk-replaced egg | 3.5 oz. | 100 g |
| 14 | Evaporated Milk, Evaporated Skim Milk, Evaporated Partly Skimmed Milk | 15.0 fl.oz. | 852 ml |
| 30.0 fl.oz. | |||
| (reconstituted to original volume) | |||
| 15 | Fish, Shell Fish | 3.5 oz. | 100 g |
| 16 | Fruits, dried | 2.0 oz. | 57 g |
| 17 | Fruits, (other than banana, lemon, lime, watermelon) | 3.5 oz. | 100 g |
| 18 | Fruits, Banana | 5.3 oz. | 150 g |
| 19 | Fruits, Lemon | 1.8 oz. | 50 g |
| 20 | Fruits, Lime | 1.8 oz. | 50 g |
| 21 | Fruits, Watermelon | 7.0 oz. | 200 g |
| 22 | Fruit Drinks, Fruit Nectars (ready to serve) | 4.0 fl.oz. | 114 ml |
| 23 | Fruit Drink Bases, Mixes and Concentrates (ready to serve) | 4.0 fl.oz. | 114 ml |
| 24 | Fruit Juices (other than lemon juice and lime juice) | 4.0 fl.oz. | 114 ml |
| 25 | Fruit Juices, Lemon | 1.0 fl.oz. | 28 ml |
| 26 | Fruit Juices, Lime | 1.0 fl.oz. | 28 ml |
| 27 | Ice Cream, Ice Milk | 3.5 oz. | 100 g |
| 28 | Infant Formulas, Prepared (ready to serve) | As directed by Label | |
| 29 | Instant Breakfast, Ready Breakfast (ready to serve) | As directed by Label | |
| 30 | Margarine | 2.0 oz. | 57 g |
| 31 | Meat Products | 3.5 oz. | 100 g |
| 32 | Meat Product Extenders | 3.5 oz. | 100 g |
| 33 | Extended Meat Products | 3.5 oz. | 100 g |
| 34 | Milk, whole | 30.0 fl.oz. | 852 ml |
| 35 | Milk Powder (reconstituted and ready to serve) | 30.0 fl.oz. | 852 ml |
| 36 | (naming the flavour) Milk | 30.0 fl.oz. | 852 ml |
| 37 | Molasses | 1.5 oz. | 43 g |
| 38 | Nuts | 1.0 oz. | 28 g |
| 39 | Peanut Butter | 1.0 oz. | 28 g |
| 40 | Poultry Products | 3.5 oz. | 100 g |
| 41 | Extended Poultry Products | 3.5 oz. | 100 g |
| 42 | Poultry Product Extenders | 3.5 oz. | 100 g |
| 43 | Simulated Meat Products excluding a simulated meat product that resembles side bacon | 3.5 oz. | 100 g |
| 44 | Simulated Poultry Products | 3.5 oz. | 100 g |
| 45 | Skim Milk, Partly Skimmed Milk | 30.0 fl.oz. | 852 ml |
| 46 | (naming the flavour) Skim Milk, (naming the flavour) Partly Skimmed Milk | 30.0 fl.oz. | 852 ml |
| 47 | Skim Milk Powder, Partly Skimmed Milk Powder (reconstituted and ready to serve) | 30.0 fl.oz. | 852 ml |
| 48 | Skim Milk with Added Milk Solids, Partly Skimmed Milk with Added Milk Solids | 30.0 fl.oz. | 852 ml |
| 49 | (naming the flavour) Skim Milk with Added Milk Solids, (naming the flavour) Partly Skimmed Milk with Added Milk Solids | 30.0 fl.oz. | 852 ml |
| 50 | Soup (ready to serve) | 7.0 fl.oz. | 200 ml |
| 51 | Sterilized Milk | 30.0 fl.oz. | 852 ml |
| 52 | Vegetable Juices | 4.0 fl.oz. | 114 ml |
| 53 | Vegetable Drinks | 4.0 fl.oz. | 114 ml |
| 54 | Vegetable Drink Concentrates, Mixes and Bases (ready to serve) | 4.0 fl.oz. | 114 ml |
| 55 | Vegetable (other than baked beans and cooked potatoes) | 3.5 oz. | 100 g |
| 56 | Vegetables, baked beans | 8.5 oz. | 250 g |
| 57 | Vegetables, cooked potatoes | 7.0 oz. | 200 g |
| 58 | Yeast | 0.5 oz. | 14 g |
| 59 | Yogurt, plain | 5.0 oz. | 150 g |
- SOR/78-64, s. 10
- SOR/84-300, s. 63(E)
APPENDICES I AND II[Repealed, SOR/81-935, s. 2]
APPENDIX IIIForms
Export Certificate
| (Under section 37 of the Food and Drugs Act) | ||
The undersigned exporter hereby certifies that the (description of article) | ||
| ||
| packaged and labelled as follows: | ||
| and marked in distinct overprinting with the word “Export” | ||
| ||
| ||
| ||
| Dated at the day of 20 . | ||
| Canada : In the matter of an Export Certificate under the Food and Drugs Act, | ||
| Province of | ||
| To Wit : | I, | |
| of the of | ||
| in the of | ||
| do solemnly declare: | ||
| ||
or | ||
| I am the of | ||
| ||
| the “Exporter” issuing the certificate above set out and have a knowledge of the matters and facts herein declared to by me (describe position of declarant as the agent of the “Exporter” in case of a Corporation issuing the certificate), | ||
| ||
| ||
| And I make this solemn declaration conscientiously believing it to be true, and knowing that it is of the same force and effect as if made under oath, and by virtue of The Canada Evidence Act. | ||
Declared before me at | ||
day of | ||
| ||
A Commissioner for Taking Oaths | ||
SCHEDULE K.1(Subsections B.01.350(1) and B.01.351(1) and (5))
Nutrition Symbols and Formats
Unilingual Horizontal Format
1(EH)  | 1(FH)  |
2(EH)  | 2(FH)  |
3(EH) 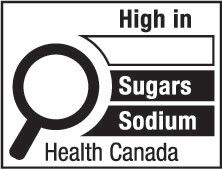 | 3(FH)  |
4(EH)  | 4(FH)  |
5(EH)  | 5(FH)  |
6(EH) 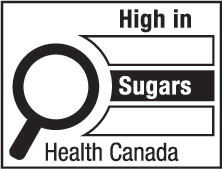 | 6(FH)  |
7(EH)  | 7(FH)  |
8(EH)  | 8(FH)  |
9(EH)  | 9(FH)  |
10(EH) 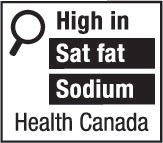 | 10(FH)  |
11(EH)  | 11(FH)  |
12(EH)  | 12(FH)  |
13(EH)  | 13(FH)  |
Unilingual Vertical Format
1(EV)  | 1(FV)  |
2(EV)  | 2(FV)  |
3(EV)  | 3(FV)  |
4(EV)  | 4(FV)  |
5(EV)  | 5(FV)  |
6(EV)  | 6(FV)  |
7(EV)  | 7(FV)  |
8(EV)  | 8(FV)  |
9(EV)  | 9(FV)  |
10(EV)  | 10(FV)  |
11(EV)  | 11(FV)  |
12(EV)  | 12(FV)  |
13(EV)  | 13(FV)  |
Bilingual Horizontal Format
1(BH)

2(BH)

3(BH)

4(BH)

5(BH)

6(BH)

7(BH)

8(BH)

9(BH)

10(BH)

11(BH)

12(BH)

13(BH)

Bilingual Vertical Format
1(BV)

2(BV)

3(BV)

4(BV)

5(BV)

6(BV)

7(BV)

8(BV)

9(BV)

10(BV)

11(BV)

12(BV)

13(BV)

SCHEDULE K.2(Subsections B.29.021(1), (2) and (8))
Supplemented Food Caution Identifier Formats
Unilingual Standard Format
ES  | FS  |
Bilingual Standard Format
BS

Bilingual Compact Format
BC

SCHEDULE L
SCHEDULE M
RELATED PROVISIONS
AMENDMENTS NOT IN FORCE
— SOR/2024-110, s. 76
76 The Food and Drug RegulationsFootnote 1 are amended by adding the following after section A.01.068:
Return to footnote 1C.R.C., c. 870
Non-application
A.01.069 These Regulations do not apply to a drug referred to in paragraph (b) or (c) of the definition biocide in subsection 1(1) of the Biocides Regulations.
— SOR/2024-110, s. 77
77 Subsection C.01.004.02(6) of the Regulations is amended by adding “and” at the end of paragraph (a), by striking out “and” at the end of paragraph (b) and by repealing paragraph (c).
— SOR/2024-110, s. 78
78 Paragraphs C.01.014.1(2)(c) and (d) of the Regulations are replaced by the following:
(c) the recommended route of administration;
— SOR/2024-110, s. 79
79 Subsection C.01.040.2(5) of the Regulations is repealed.
— SOR/2024-110, s. 80
80 The definition antimicrobial agent in subsection C.01A.001(1) of the Regulations is repealed.
— SOR/2024-110, s. 81
81 Subsection C.01A.002(1) of the Regulations is amended by adding “and” at the end of paragraph (c), by striking out “and” at the end of paragraph (d) and by repealing paragraph (e).
— SOR/2024-110, s. 82
82 Section C.02.002.1 of the Regulations is repealed.
— SOR/2024-136, s. 1
1 Section C.01.051 of the Food and Drug RegulationsFootnote 1 and the heading before it are replaced by the following:
Return to footnote 1C.R.C., c. 870
Recall Reporting
C.01.051 (1) If a manufacturer who sells a drug in dosage form or an active ingredient or a person who imports into and sells a drug in dosage form or an active ingredient in Canada decides to recall any of those drugs without being ordered to do so by the Minister, the manufacturer or importer shall, within 24 hours after making the decision, provide the Minister with the following information in writing:
(a) the drug’s proper name or the common name, if there is no proper name;
(b) an indication as to whether the drug is a drug in dosage form or an active ingredient;
(c) in the case of a drug in dosage form,
(i) the brand name,
(ii) the drug identification number assigned under subsection C.01.014.2(1),
(iii) the dosage form,
(iv) the strength,
(v) the names of the persons in Canada, other than consumers that purchased the drug at the retail level, to whom the drug was sold by the manufacturer or importer and the quantity sold to each of the named persons, and
(vi) an assessment of the effect that the recall may have on the manufacturer’s or importer’s ability to meet demand for the drug in Canada;
(d) the lot numbers of the drug;
(e) the dates of fabrication of the drug;
(f) the expiration dates of the drug;
(g) the quantity of the drug that was fabricated in Canada;
(h) the quantity of the drug that was imported;
(i) the quantity of the drug that the manufacturer or importer sold to persons in Canada and the period during which it was sold;
(j) the quantity of the drug that the manufacturer or importer exported, as well as the quantity exported, by country;
(k) the quantity of the drug in Canada that is in the possession or control of the manufacturer or importer;
(l) an assessment of the risk of injury to human health posed by the drug, including because of a failure of its effectiveness;
(m) the names and civic addresses of the manufacturer and fabricator of the drug and of any importers of the drug;
(n) the name and contact information of the individual who is responsible for the recall;
(o) the expected dates for the start and completion of the recall; and
(p) the reasons for the recall and the date on which and manner in which the situation that prompted the recall was discovered.
(2) The manufacturer or importer shall
(a) before starting the recall, provide the Minister with a copy of any communications that the manufacturer or importer intends to use in connection with the start of the recall; and
(b) after starting the recall, provide the Minister, on request and within the time specified by the Minister, with a copy of any additional communications that the manufacturer or importer uses, or intends to use, in connection with the recall.
(3) The manufacturer or importer shall, within 72 hours after making the decision to recall the drug, provide the Minister with the following information in writing:
(a) the strategy for conducting the recall, including the time and manner in which the Minister will be informed of the progress of the recall; and
(b) a description of the measures that are intended to be taken to prevent a recurrence of the situation that prompted the recall.
(4) The manufacturer or importer shall, within 30 days after completing the recall, provide the Minister with the following information in writing:
(a) the results of the recall; and
(b) a description of the measures that have been or will be taken to prevent a recurrence of the situation that prompted the recall.
(5) In this section and in section C.01.051.1, active ingredient and fabricate have the same meaning as in subsection C.01A.001(1).
C.01.051.1 (1) A person who is ordered by the Minister under section 21.3 of the Act to recall a drug in dosage form, or an active ingredient, shall provide the Minister with the following information in the time and manner specified by the Minister:
(a) the quantity of the drug that has been sold by the person at the retail level to consumers in Canada;
(b) if the person has sold the drug to persons in Canada other than consumers referred to in paragraph (a),
(i) the names of those persons and the quantity that has been sold to each of them, and
(ii) the period during which the drug was sold to those persons;
(c) the quantity of the drug that the person has exported from Canada, as well as the quantity exported by country;
(d) the quantity of the drug in Canada that is in the possession or control of the person;
(e) the name, title and contact information of an individual from whom the Minister may obtain additional information concerning the recall;
(f) the strategy for conducting the recall;
(g) any other information that the Minister has reasonable grounds to believe is necessary to mitigate the risk of injury to human health; and
(h) if the person is in a position to prevent a recurrence of the situation that prompted the recall, a description of the measures that they intend to take to prevent a recurrence.
(2) The person shall notify the Minister without delay of any change to the information referred to in paragraph (1)(e).
(3) The person shall
(a) before starting the recall, provide the Minister with a copy of any communications that the person intends to use in connection with the start of the recall; and
(b) after starting the recall, provide the Minister with, on request and within the time specified by the Minister, a copy of any additional communications that the person uses, or intends to use, in connection with the recall.
(4) The person shall notify the Minister in writing, within 24 hours, of the start and completion of the recall.
(5) The person shall, within 30 days after completing the recall, provide the Minister with the following information in writing:
(a) the results of the recall; and
(b) if the person is in a position to prevent a recurrence of the situation that prompted the recall, a description of the measures that they have taken or will take to prevent a recurrence.
— SOR/2024-136, s. 3
3 Paragraph C.02.012(1)(a) of the French version of the Regulations is replaced by the following:
a) un système de contrôle qui permet le rappel rapide et complet de tout lot ou tout lot de fabrication de la drogue se trouvant sur le marché;
— SOR/2024-136, s. 5
5 (1) Subsection C.02.022(1) of the French version of the Regulations is replaced by the following:
C.02.022 (1) Le grossiste, le distributeur visé à l’article C.01A.003 et l’importateur d’une drogue sous forme posologique conservent les dossiers sur la vente de chaque lot ou lot de fabrication de la drogue pendant un an après sa date limite d’utilisation afin de pouvoir en faire le rappel, à moins que la licence d’établissement de l’intéressé ne prévoie une autre période.
(2) The portion of subsection C.02.022(2) of the French version of the Regulations before paragraph (a) is replaced by the following:
(2) Le distributeur d’un ingrédient actif visé à l’alinéa C.01A.003a) et le grossiste et l’importateur d’un ingrédient actif conservent les dossiers sur la vente de chaque lot ou lot de fabrication de l’ingrédient actif pendant celle des périodes ci-après qui s’applique afin de pouvoir en faire le rappel, à moins que l’intéressé ne détienne une licence d’établissement qui prévoit une autre période :
- Date modified: